
Emerging Biomarkers and Innovative Therapeutic Strategies in Diabetic Kidney Disease: A Pathway to Precision Medicine



Abstract
1. Introduction
2. Pathobiology of Diabetic Nephropathy
3. Structural Changes in the Kidney
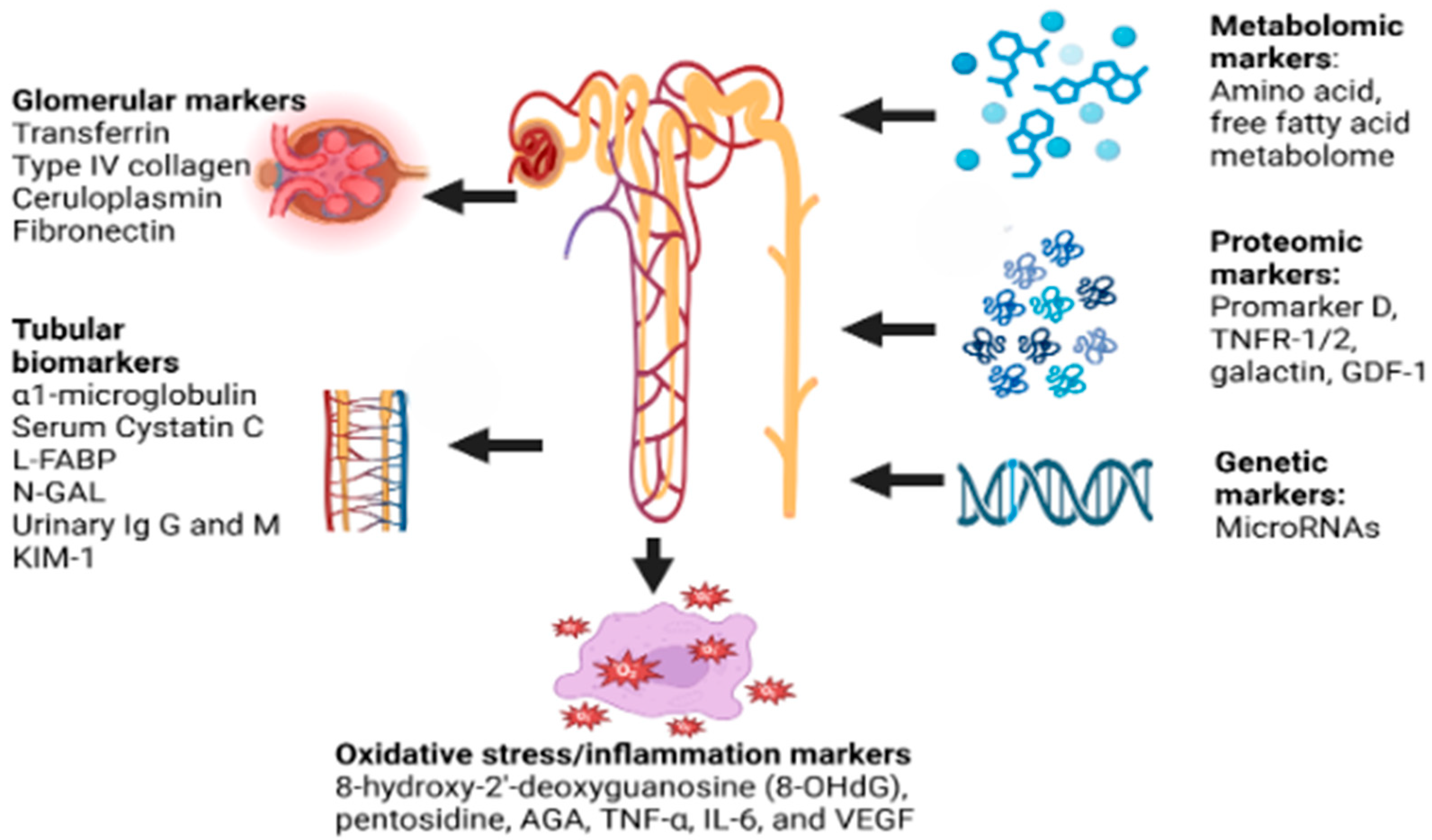
4. Evaluation
5. Glomerular Markers
6. Tubular Biomarkers
7. Metabolomics
8. Genetic Markers
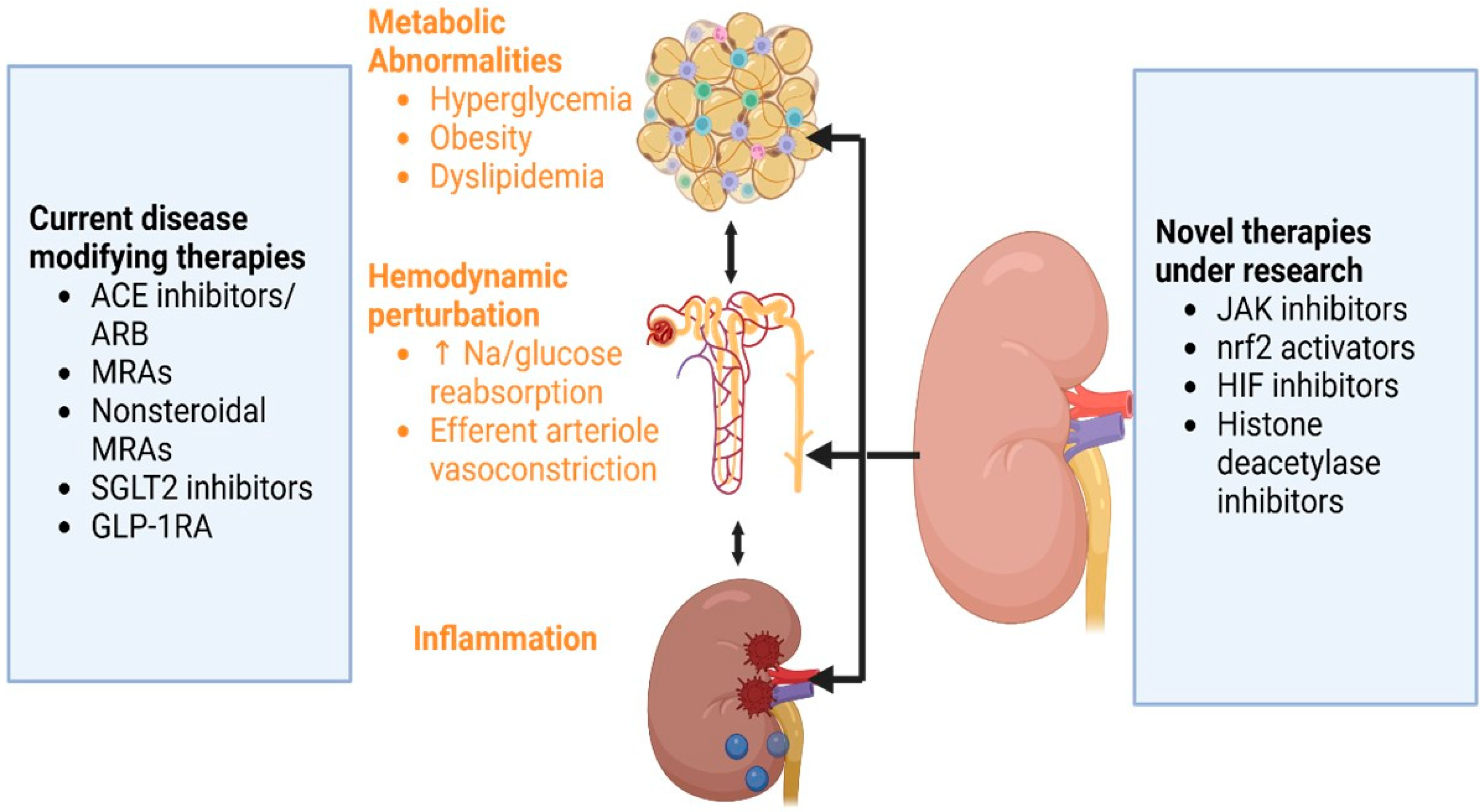
9. Current Disease-Modifying Approaches for DKD
10. Clinical Studies on Emerging Therapies in DKD
11. Future Therapeutic Targets and Drug Development
12. Conclusions
Funding
Conflicts of Interest
References
- GBD 2016 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1260–1344. [Google Scholar]
- Francis, A.; Harhay, M.N.; Ong, A.C.M.; Tummalapalli, S.L.; Ortiz, A.; Fogo, A.B.; Fliser, D.; Roy-Chaudhury, P.; Fontana, M.; Nangaku, M.; et al. Chronic kidney disease and the global public health agenda: An international consensus. Nat. Rev. Nephrol. 2024, 20, 473–485. [Google Scholar] [PubMed]
- GBD 2017 DALYs and HALE Collaborators. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar]
- Naaman, S.C.; Bakris, G.L. Diabetic Nephropathy: Update on Pillars of Therapy Slowing Progression. Diabetes Care 2023, 46, 1574–1586. [Google Scholar]
- Cheng, H.T.; Xu, X.; Lim, P.S.; Hung, K.Y. Worldwide Epidemiology of Diabetes-Related End-Stage Renal Disease, 2000–2015. Diabetes Care 2020, 44, 89–97. [Google Scholar]
- Patel, D.M.; Bose, M.; Cooper, M.E. Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 2218. [Google Scholar] [CrossRef]
- Ricciardi, C.A.; Gnudi, L. Kidney disease in diabetes: From mechanisms to clinical presentation and treatment strategies. Metabolism 2021, 124, 154890. [Google Scholar]
- Hu, Q.; Chen, Y.; Deng, X.; Li, Y.; Ma, X.; Zeng, J.; Zhao, Y. Diabetic nephropathy: Focusing on pathological signals, clinical treatment, and dietary regulation. Biomed. Pharmacother. 2023, 159, 114252. [Google Scholar]
- Loeffler, I.; Wolf, G. Mechanisms of Interstitial Fibrosis in Diabetic Nephropathy. In Diabetic Nephropathy: Pathophysiology and Clinical Aspects [Internet]; Roelofs, J.J., Vogt, L., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 227–251. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar]
- Cau, S.B.; Bruder-Nascimento, A.; Silva, M.B.; Ramalho, F.N.Z.; Mestriner, F.; Alves-Lopes, R.; Ferreira, N.; Tostes, R.C.; Bruder-Nascimento, T. Angiotensin-II activates vascular inflammasome and induces vascular damage. Vasc. Pharmacol. 2021, 139, 106881. [Google Scholar]
- Koszegi, S.; Molnar, A.; Lenart, L.; Hodrea, J.; Balogh, D.B.; Lakat, T.; Szkibinszkij, E.; Hosszu, A.; Sparding, N.; Genovese, F.; et al. RAAS inhibitors directly reduce diabetes-induced renal fibrosis via growth factor inhibition. J. Physiol. 2018, 597, 193–209. [Google Scholar] [PubMed]
- Rincon-Choles, H.; Kasinath, B.S.; Gorin, Y.; Abboud, H.E. Angiotensin II and growth factors in the pathogenesis of diabetic nephropathy. Kidney Int. 2002, 62, S8–S11. [Google Scholar]
- Wu, T.; Ding, L.; Andoh, V.; Zhang, J.; Chen, L. The Mechanism of Hyperglycemia-Induced Renal Cell Injury in Diabetic Nephropathy Disease: An Update. Life 2023, 13, 539. [Google Scholar] [CrossRef]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Moreno, J.A.; Ruiz-Ortega, M.; Ramos, A.M.; Sanz, A.B.; Ortiz, A. Downregulation of kidney protective factors by inflammation: Role of transcription factors and epigenetic mechanisms. Am. J. Physiol. Ren. Physiol. 2016, 311, F1329–F1340. [Google Scholar]
- Mohandes, S.; Doke, T.; Hu, H.; Mukhi, D.; Dhillon, P.; Susztak, K. Molecular pathways that drive diabetic kidney disease. J. Clin. Investig. 2023, 133, e165654. [Google Scholar]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [PubMed]
- Hayashi, K. Altered DNA methylation in kidney disease: Useful markers and therapeutic targets. Clin. Exp. Nephrol. 2022, 26, 309–315. [Google Scholar]
- Cefalu, W.T.; Rodgers, G.P. Diabetes control and complications trial/epidemiology of diabetes interventions and complications study: Continuing to build on 40 years of diabetes research. Diabetes Care 2024, 47, 1518–1521. [Google Scholar]
- Thomas, M.C. Targeting the Pathobiology of Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2021, 28, 282–289. [Google Scholar]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar]
- Marshall, C.B. Rethinking glomerular basement membrane thickening in diabetic nephropathy: Adaptive or pathogenic? Am. J. Physiol. Ren. Physiol. 2016, 311, F831–F843. [Google Scholar]
- Ziyadeh, F.N.; Wolf, G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr. Diabetes Rev. 2008, 4, 39–45. [Google Scholar]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primer 2015, 1, 15018. [Google Scholar]
- Gilbert, R.E. Proximal tubulopathy: Prime mover and key therapeutic target in diabetic kidney disease. Diabetes 2017, 66, 791–800. [Google Scholar] [PubMed]
- Moresco, R.N.; Bochi, G.V.; Stein, C.S.; De Carvalho, J.A.M.; Cembranel, B.M.; Bollick, Y.S. Urinary kidney injury molecule-1 in renal disease. Clin. Chim. Acta 2018, 487, 15–21. [Google Scholar]
- Najafian, B.; Kim, Y.; Crosson, J.T.; Mauer, M. Atubular Glomeruli and Glomerulotubular Junction Abnormalities in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, 908. [Google Scholar]
- Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Fernandez-Fernandez, B.; Mora-Fernández, C.; Marchant, V.; Donate-Correa, J.; Navarro-González, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Int. 2023, 103, 282–296. [Google Scholar]
- Yamanouchi, M.; Furuichi, K.; Hoshino, J.; Ubara, Y.; Wada, T. Nonproteinuric diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 573–581. [Google Scholar]
- Barutta, F.; Bellini, S.; Canepa, S.; Durazzo, M.; Gruden, G. Novel biomarkers of diabetic kidney disease: Current status and potential clinical application. Acta Diabetol. 2021, 58, 819–830. [Google Scholar]
- Currie, G.; McKay, G.; Delles, C. Biomarkers in diabetic nephropathy: Present and future. World J. Diabetes 2014, 5, 763–776. [Google Scholar]
- Żyłka, A.; Dumnicka, P.; Kuśnierz-Cabala, B.; Gala-Błądzińska, A.; Ceranowicz, P.; Kucharz, J.; Ząbek-Adamska, A.; Maziarz, B.; Drożdż, R.; Kuźniewski, M. Markers of glomerular and tubular damage in the early stage of kidney disease in type 2 diabetic patients. Mediat. Inflamm. 2018, 2018, 7659243. [Google Scholar]
- Wang, C.; Li, C.; Gong, W.; Lou, T. New urinary biomarkers for diabetic kidney disease. Biomark. Res. 2013, 1, 9. [Google Scholar]
- Dağdeviren Çakır, A.; Saygılı, S.K.; Canpolat, N.; Konukoğlu, D.; Turan, H.; Çalışkan, S.; Sever, L.; Ercan, O.; Evliyaoğlu, O. Elevated Urinary VEGF-A, Transferrin, and Angiotensinogen Levels in Normoalbuminuric Children and Adolescents with Type 1 Diabetes: Can They Be Early Markers of Diabetic Kidney Disease? Horm. Res. Paediatr. 2021, 94, 426–432. [Google Scholar] [PubMed]
- Araki, S.I.; Haneda, M.; Koya, D.; Isshiki, K.; Kume, S.; Sugimoto, T.; Kawai, H.; Nishio, Y.; Kashiwagi, A.; Uzu, T.; et al. Association between urinary type IV collagen level and deterioration of renal function in type 2 diabetic patients without overt proteinuria. Diabetes Care 2010, 33, 1805–1810. [Google Scholar] [PubMed]
- MacIsaac, R.J.; Ekinci, E.I.; Jerums, G. Markers of and Risk Factors for the Development and Progression of Diabetic Kidney Disease. Am. J. Kidney Dis. 2014, 63, S39–S62. [Google Scholar] [PubMed]
- Bonventre, J.V. Can We Target Tubular Damage to Prevent Renal Function Decline in Diabetes? Semin. Nephrol. 2012, 32, 452–462. [Google Scholar]
- Gudehithlu, K.P.; Garcia-Gomez, I.; Vernik, J.; Brecklin, C.; Kraus, M.; Cimbaluk, D.J.; Hart, P.; Dunea, G.; Arruda, J.A.; Singh, A.K. In Diabetic Kidney Disease Urinary Exosomes Better Represent Kidney Specific Protein Alterations Than Whole Urine. Am. J. Nephrol. 2016, 42, 418–424. [Google Scholar]
- Li, X.; Miao, Y.; Li, T.; Liu, X.; Xu, L.; Guo, J.; Yu, X.; Sun, B.; Zhu, Y.; Ai, D.; et al. Integrin β6 mediates epithelial–mesenchymal transition in diabetic kidney disease. Mol. Cell. Endocrinol. 2023, 572, 111955. [Google Scholar]
- Zhou, Y.; Zhang, Y.; Chen, J.; Wang, T.; Li, H.; Wu, F.; Shang, J.; Zhao, Z. Diagnostic value of α1-MG and URBP in early diabetic renal impairment. Front. Physiol. 2023, 14, 1173982. [Google Scholar]
- Fiseha, T. Urinary biomarkers for early diabetic nephropathy in type 2 diabetic patients. Biomark. Res. 2015, 3, 1–7. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, Y.; Xue, C. Diagnostic value of serum cystatin C for diabetic nephropathy: A meta-analysis. BMC Endocr. Disord. 2022, 22, 149. [Google Scholar]
- Rico-Fontalvo, J.; Aroca-Martínez, G.; Daza-Arnedo, R.; Cabrales, J.; Rodríguez-Yanez, T.; Cardona-Blanco, M.; Montejo-Hernández, J.; Barrios, D.R.; Patiño-Patiño, J.; Rodríguez, E.O. Novel Biomarkers of Diabetic Kidney Disease. Biomolecules 2023, 13, 633. [Google Scholar]
- Greco, M.; Chiefari, E.; Mirabelli, M.; Salatino, A.; Tocci, V.; Cianfrone, P.; Foti, D.P.; Brunetti, A. Plasma or Urine Neutrophil Gelatinase-Associated Lipocalin (NGAL): Which Is Better at Detecting Chronic Kidney Damage in Type 2 Diabetes? Endocrines 2022, 3, 175–186. [Google Scholar] [CrossRef]
- Mishra, A.; Pant, N.; Andriyas, E.A.; Umar, M.H.; Saxena, A.K.; Hussain, I.; Kushwaha, N. Novel Biomarkers in Early Prediction of Diabetic Nephropathy: A Systematic Review. World J. Adv. Res. Rev. 2024, 23, 1349–1355. [Google Scholar]
- Zhao, X.; Chen, X.; Zhang, Y.; George, J.; Cobbs, A.; Wang, G.; Li, L.; Emmett, N. Kidney Injury Molecule-1 Is Upregulated in Renal Lipotoxicity and Mediates Palmitate-Induced Tubular Cell Injury and Inflammatory Response. Int. J. Mol. Sci. 2019, 20, 3406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, M.; Cheng, C.K.; Li, Q. Tubular injury in diabetic kidney disease: Molecular mechanisms and potential therapeutic perspectives. Front. Endocrinol. 2023, 14, 1238927. [Google Scholar]
- Huo, W.; Zhang, K.; Nie, Z.; Li, Q.; Jin, F. Kidney injury molecule-1 (KIM-1): A novel kidney-specific injury molecule playing potential double-edged functions in kidney injury. Transplant. Rev. 2010, 24, 143–146. [Google Scholar]
- Zakiyanov, O.; Kalousová, M.; Zima, T.; Tesař, V. Chapter Four—Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in kidney disease. In Advances in Clinical Chemistry [Internet]; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 141–212. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0065242321000147 (accessed on 20 February 2025).
- Han, Y.Z.; Du, B.X.; Zhu, X.Y.; Wang, Y.Z.; Zheng, H.J.; Liu, W.J. Lipid metabolism disorder in diabetic kidney disease. Front. Endocrinol. 2024, 15, 1336402. [Google Scholar]
- Shaker, A.M.; Mohamed, M.E.; Ramzy, T.; Ali, M.I. Serum fatty acid-binding protein 4 as a biomarker for early detection of diabetic nephropathy in type 2 diabetes. Egypt. J. Intern. Med. 2023, 35, 22. [Google Scholar]
- Zhang, Y.; Zhang, S.; Wang, G. Metabolomic biomarkers in diabetic kidney diseases—A systematic review. J. Diabetes Complicat. 2015, 29, 1345–1351. [Google Scholar]
- Chen, J.; Zhang, Q.; Liu, D.; Liu, Z. Exosomes: Advances, development and potential therapeutic strategies in diabetic nephropathy. Metabolism 2021, 122, 154834. [Google Scholar] [PubMed]
- Dieter, C.; Assmann, T.S.; Costa, A.R.; Canani, L.H.; De Souza, B.M.; Bauer, A.C.; Crispim, D. MiR-30e-5p and MiR-15a-5p expressions in plasma and urine of type 1 diabetic patients with diabetic kidney disease. Front. Genet. 2019, 10, 563. [Google Scholar]
- Dong, W.; Zhang, H.; Zhao, C.; Luo, Y.; Chen, Y. Silencing of miR-150-5p ameliorates diabetic nephropathy by targeting SIRT1/p53/AMPK pathway. Front. Physiol. 2021, 12, 624989. [Google Scholar]
- Arslan, G.; Karabulut, Y.Y.; Yeleser, İ.; Erdal, M.E.; Demir, S.; Özdemir, A.A. Correlation of hsa-mirna-342–3p and SOX 6 Expression with Diabetic Nephropathy Classification, Prognostic Histomorphological Parameters and Laboratory Findings in Diabetic Nephropathy. Ann. Diagn. Pathol. 2025, 76, 152461. [Google Scholar] [PubMed]
- Zhang, M.; Lu, Y.; Wang, L.; Mao, Y.; Hu, X.; Chen, Z. Autocrine small extracellular vesicles induce tubular phenotypic transformation in diabetic nephropathy via miR-21-5p. Gene 2025, 938, 149156. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Su, R.; Zhen, J.; Liu, X.; Liu, G. Small extracellular vesicles-shuttled miR-23a-3p from mesenchymal stem cells alleviate renal fibrosis and inflammation by inhibiting KLF3/STAT3 axis in diabetic kidney disease. Int. Immunopharmacol. 2024, 139, 112667. [Google Scholar]
- Sharma, V.; Khokhar, M.; Panigrahi, P.; Gadwal, A.; Setia, P.; Purohit, P. Advancements, Challenges, and clinical implications of integration of metabolomics technologies in diabetic nephropathy. Clin. Chim. Acta. 2024, 561, 119842. [Google Scholar]
- Pena, M.J.; Lambers Heerspink, H.J.; Hellemons, M.E.; Friedrich, T.; Dallmann, G.; Lajer, M.; Bakker, S.J.L.; Gansevoort, R.T.; Rossing, P.; de Zeeuw, D.; et al. Urine and plasma metabolites predict the development of diabetic nephropathy in individuals with Type 2 diabetes mellitus. Diabet. Med. 2014, 31, 1138–1147. [Google Scholar]
- Pereira, P.R.; Carrageta, D.F.; Oliveira, P.F.; Rodrigues, A.; Alves, M.G.; Monteiro, M.P. Metabolomics as a tool for the early diagnosis and prognosis of diabetic kidney disease. Med. Res. Rev. 2022, 42, 1518–1544. [Google Scholar]
- Barr, S.I.; Abd El-Azeem, E.M.; Bessa, S.S.; Mohamed, T.M. Association of serum uromodulin with diabetic kidney disease: A systematic review and meta-analysis. BMC Nephrol. 2024, 25, 421. [Google Scholar] [CrossRef]
- Budge, K.; Dellepiane, S.; Yu, S.M.; Cravedi, P. Complement, a Therapeutic Target in Dia-betic Kidney Disease. Front. Med. 2021, 7, 599236. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Schnellmann, R.G. Pharmacological Targeting of Mitochondria in Diabetic Kidney Disease. Pharmacol. Rev. 2023, 75, 250–262. [Google Scholar] [CrossRef]
- Das, S.; Devi Rajeswari, V.; Venkatraman, G.; Elumalai, R.; Dhanasekaran, S.; Ramanathan, G. Current updates on metabolites and its interlinked pathways as biomarkers for diabetic kidney disease: A systematic review. Transl. Res. 2024, 265, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pezzolesi, M.G. Advances in understanding the genetic basis of diabetic kidney disease. Acta Diabetol. 2018, 55, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Winkler, T.W.; Rasheed, H.; Teumer, A.; Gorski, M.; Rowan, B.X.; Stanzick, K.J.; Thomas, L.F.; Tin, A.; Hoppmann, A.; Chu, A.Y.; et al. Differential and shared genetic effects on kidney function between diabetic and non-diabetic individuals. Commun. Biol. 2022, 5, 580. [Google Scholar] [CrossRef] [PubMed]
- Sandholm, N.; Dahlström, E.H.; Groop, P.H. Genetic and epigenetic background of diabetic kidney disease. Front. Endocrinol. 2023, 14, 1163001. [Google Scholar] [CrossRef]
- Thompson, M.; Hill, B.L.; Rakocz, N.; Chiang, J.N.; Geschwind, D.; Sankararaman, S.; Hofer, I.; Cannesson, M.; Zaitlen, N.; Halperin, E. Methylation risk scores are associated with a collection of phenotypes within electronic health record systems. NPJ Genom. Med. 2022, 7, 50. [Google Scholar] [CrossRef]
- Jia, Y.; Reddy, M.A.; Das, S.; Oh, H.J.; Abdollahi, M.; Yuan, H.; Zhang, E.; Lanting, L.; Wang, M.; Natarajan, R. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1–induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 2019, 294, 12695–12707. [Google Scholar] [CrossRef]
- Gæde, P.; Lund-Andersen, H.; Parving, H.H.; Pedersen, O. Effect of a Multifactorial Intervention on Mortality in Type 2 Diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef]
- Ueki, K.; Sasako, T.; Okazaki, Y.; Kato, M.; Okahata, S.; Katsuyama, H.; Haraguchi, M.; Morita, A.; Ohashi, K.; Hara, K.; et al. Effect of an intensified multifactorial intervention on cardiovascular outcomes and mortality in type 2 diabetes (J-DOIT3): An open-label, randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 951–964. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The Effect of Angiotensin-Converting-Enzyme Inhibition on Diabetic Nephropathy. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of Losartan on Renal and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Lehnert, H.; Bröchner-Mortensen, J.; Gomis, R.; Andersen, S.; Arner, P. Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N. Engl. J. Med. 2001, 345, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Viberti, G.; Wheeldon, N.M. Microalbuminuria Reduction With Valsartan in Patients With Type 2 Diabetes Mellitus. Circulation 2002, 106, 672–678. [Google Scholar] [CrossRef]
- Makino, H.; Haneda, M.; Babazono, T.; Moriya, T.; Ito, S.; Iwamoto, Y.; Kawamori, R.; Takeuchi, M.; Katayama, S. Prevention of transition from incipient to overt nephropathy with telmisartan in patients with type 2 diabetes. Diabetes Care 2007, 30, 1577–1578. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Schmieder, R.E.; McQueen, M.; Dyal, L.; Schumacher, H.; Pogue, J.; Wang, X.; Maggioni, A.; Budaj, A.; Chaithiraphan, S.; et al. ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): A multicentre, randomised, double-blind, controlled trial. Lancet 2008, 372, 547–553. [Google Scholar] [CrossRef]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined Angiotensin Inhibition for the Treatment of Diabetic Nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef]
- Hou, J.; Xiong, W.; Cao, L.; Wen, X.; Li, A. Spironolactone Add-on for Preventing or Slowing the Progression of Diabetic Nephropathy: A Meta-analysis. Clin. Ther. 2015, 37, 2086–2103.e10. [Google Scholar] [CrossRef]
- Sarafidis, P.; Iatridi, F.; Ferro, C.; Alexandrou, M.E.; Fernandez-Fernandez, B.; Kanbay, M.; Mallamaci, F.; Nistor, I.; Rossignol, P.; Wanner, C.; et al. Mineralocorticoid receptor antagonist use in chronic kidney disease with type 2 diabetes: A clinical practice document by the European Renal Best Practice (ERBP) board of the European Renal Association (ERA). Clin. Kidney J. 2023, 16, 1885–1907. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Girerd, S.; Jaisser, F. Mineralocorticoid receptor antagonists and kidney diseases: Pathophysiological basis. Kidney Int. 2019, 96, 302–319. [Google Scholar] [CrossRef]
- Tesch, G.H.; Young, M.J. Mineralocorticoid Receptor Signaling as a Therapeutic Target for Renal and Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, C. Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression. Int. J. Mol. Sci. 2024, 25, 3086. [Google Scholar] [CrossRef]
- Williams, G.H.; Burgess, E.; Kolloch, R.E.; Ruilope, L.M.; Niegowska, J.; Kipnes, M.S.; Roniker, B.; Patrick, J.L.; Krause, S.L. Efficacy of eplerenone versus enalapril as monotherapy in systemic hypertension. Am. J. Cardiol. 2004, 93, 990–996. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Filippatos, G.; Nowack, C.; Kolkhof, P.; Joseph, A.; Mentenich, N.; Pitt, B.; et al. Design and Baseline Characteristics of the Finerenone in Reducing Cardiovascular Mortality and Morbidity in Diabetic Kidney Disease Trial. Am. J. Nephrol. 2019, 50, 345–356. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Nowack, C.; Kolkhof, P.; Ferreira, A.C.; Schloemer, P.; Filippatos, G. FIDELIO-DKD study investigators. Design and baseline characteristics of the finerenone in reducing kidney failure and disease progression in diabetic kidney disease trial. Am. J. Nephrol. 2019, 50, 333–344. [Google Scholar] [CrossRef]
- Phongphithakchai, A.; Tedasen, A.; Netphakdee, R.; Leelawattana, R.; Srithongkul, T.; Raksasuk, S.; Huang, J.C.; Chatatikun, M. Dapagliflozin in Chronic Kidney Disease: Insights from Network Pharmacology and Molecular Docking Simulation. Life 2025, 15, 437. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Sugiura, Y.; Saito, H.; Sugahara, M.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Suematsu, M.; Nangaku, M.; Tanaka, T. Sodium–glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int. 2018, 94, 912–925. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: Biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 2021, 17, 227–244. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Prato, S.D.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Shaman, A.M.; Bain, S.C.; Bakris, G.L.; Buse, J.B.; Idorn, T.; Mahaffey, K.W.; Mann, J.F.; Nauck, M.A.; Rasmussen, S.; Rossing, P.; et al. Effect of the glucagon-like peptide-1 receptor agonists semaglutide and liraglutide on kidney outcomes in patients with type 2 diabetes: Pooled analysis of SUSTAIN 6 and LEADER. Circulation 2022, 145, 575–585. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef]
- Singh, A.K.; Farag, Y.M.K.; Zheng, Z.; Bakris, G.L. Clinical trial designs of emerging therapies for diabetic kidney disease (DKD). Postgrad Med. 2024, 136, 585–593. [Google Scholar] [CrossRef]
- Herrington, W.G.; Baigent, C.; Haynes, R. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 2301–2302. [Google Scholar]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Rossing, P.; Sjöström, C.D.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Rossing, P.; Baeres, F.M.M.; Bakris, G.; Bosch-Traberg, H.; Gislum, M.; Gough, S.C.L.; Idorn, T.; Lawson, J.; Mahaffey, K.W.; Mann, J.F.E.; et al. The rationale, design and baseline data of FLOW, a kidney outcomes trial with once-weekly semaglutide in people with type 2 diabetes and chronic kidney disease. Nephrol. Dial. Transplant. 2023, 38, 2041–2051. [Google Scholar] [CrossRef]
- Rossing, P.; Anker, S.D.; Filippatos, G.; Pitt, B.; Ruilope, L.M.; Birkenfeld, A.L.; McGill, J.B.; Rosas, S.E.; Joseph, A.; Gebel, M.; et al. Finerenone in Patients With Chronic Kidney Disease and Type 2 Diabetes by Sodium–Glucose Cotransporter 2 Inhibitor Treatment: The FIDELITY Analysis. Diabetes Care 2022, 45, 2991–2998. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Heerspink, H.J.L.; Vart, P.; Claggett, B.L.; Fletcher, R.A.; Arnott, C.; Costa, J.d.O.; Falster, M.O.; Pearson, S.-A.; Mahaffey, K.W.; et al. Estimated Lifetime Cardiovascular, Kidney, and Mortality Benefits of Combination Treatment With SGLT2 Inhibitors, GLP-1 Receptor Agonists, and Nonsteroidal MRA Compared With Conventional Care in Patients With Type 2 Diabetes and Albuminuria. Circulation 2024, 149, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Petrazzuolo, A.; Sabiu, G.; Assi, E.; Maestroni, A.; Pastore, I.; Lunati, M.E.; Montefusco, L.; Loretelli, C.; Rossi, G.; Nasr, M.B.; et al. Broadening horizons in mechanisms, management, and treatment of diabetic kidney disease. Pharmacol. Res. 2023, 190, 106710. [Google Scholar] [CrossRef]
- Heiss, E.H.; Schachner, D.; Werner, E.R.; Dirsch, V.M. Active NF-E2-related Factor (Nrf2) Contributes to Keep Endothelial NO Synthase (eNOS) in the Coupled State. J. Biol. Chem. 2009, 284, 31579–31586. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic Potential of Nrf2 Activators in Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef]
- Conde, E.; Giménez-Moyano, S.; Martín-Gómez, L.; Rodríguez, M.; Ramos, M.E.; Aguado-Fraile, E.; Blanco-Sanchez, I.; Saiz, A.; García-Bermejo, M.L. HIF-1α induction during reperfusion avoids maladaptive repair after renal ischemia/reperfusion involving miR127-3p. Sci. Rep. 2017, 7, 41099. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef]
- Zhao, M.; Cao, Y.; Ma, L. New insights in the treatment of DKD: Recent advances and future prospects. BMC Nephrol. 2025, 26, 72. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Biomarker | Sensitivity (%) | Specificity (%) | Role in DKD |
|---|---|---|---|
| Serum NGAL | 79 | 61 | Early biomarker of DKD, reflecting tubular injury and inflammation |
| Urine NGAL | 80 | 61 | Early tubular injury marker, elevated before albuminuria |
| Serum Cystatin C | 68 | 90 | Estimation of kidney function |
| Urine L-FABP | 98 | 90 | Reflects tubular damage and oxidative stress |
| Urine NGAL/Creatinine | 60 | 87 | Early tubular injury in DKD, aiding in the detection of kidney damage |
| Serum Uromodulin | 95 | 43 | Tubular health indicator, inversely related to DKD progression |
| Urine KIM-1 | 79 | 51 | Early tubular injury marker, associated with DKD progression |
| Urine Transferrin | 47 | 98 | Early glomerular injury marker, detected before albuminuria |
| Urine IgG | 74 | 93 | Glomerular permeability dysfunction in DKD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetty, S.; Suvarna, R.; Awasthi, A.; Bhojaraja, M.V.; Pappachan, J.M. Emerging Biomarkers and Innovative Therapeutic Strategies in Diabetic Kidney Disease: A Pathway to Precision Medicine. Diagnostics 2025, 15, 973. https://doi.org/10.3390/diagnostics15080973
Shetty S, Suvarna R, Awasthi A, Bhojaraja MV, Pappachan JM. Emerging Biomarkers and Innovative Therapeutic Strategies in Diabetic Kidney Disease: A Pathway to Precision Medicine. Diagnostics. 2025; 15(8):973. https://doi.org/10.3390/diagnostics15080973
Chicago/Turabian StyleShetty, Sahana, Renuka Suvarna, Avivar Awasthi, Mohan V. Bhojaraja, and Joseph M. Pappachan. 2025. "Emerging Biomarkers and Innovative Therapeutic Strategies in Diabetic Kidney Disease: A Pathway to Precision Medicine" Diagnostics 15, no. 8: 973. https://doi.org/10.3390/diagnostics15080973
APA StyleShetty, S., Suvarna, R., Awasthi, A., Bhojaraja, M. V., & Pappachan, J. M. (2025). Emerging Biomarkers and Innovative Therapeutic Strategies in Diabetic Kidney Disease: A Pathway to Precision Medicine. Diagnostics, 15(8), 973. https://doi.org/10.3390/diagnostics15080973