A Rapid and Reliable Test for BRCA1 Promoter Hypermethylation in Paraffin Tissue Using Pyrosequencing

Abstract
1. Introduction
2. Materials and Methods
3. Results
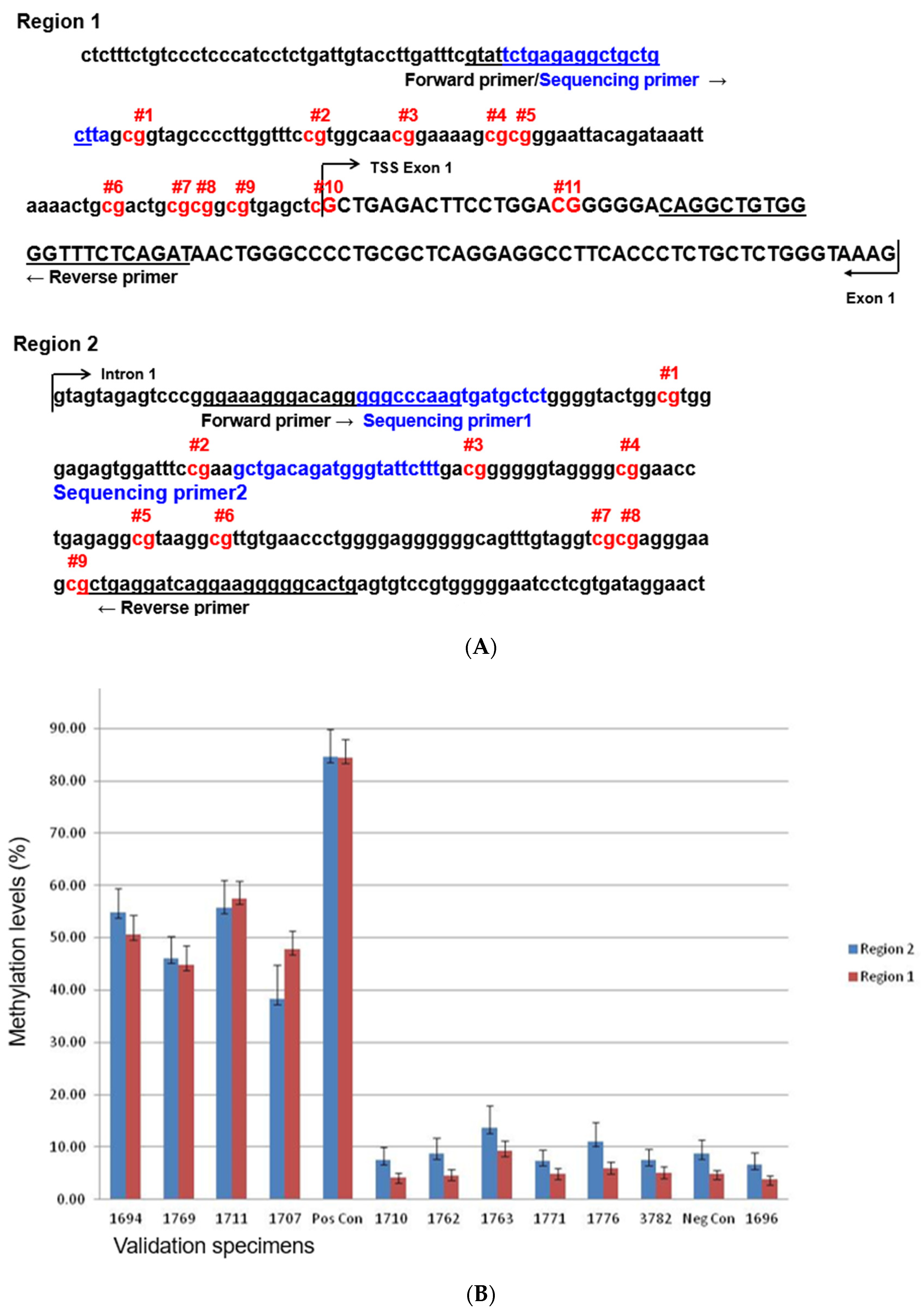
3.1. Design and Interpretation of the Pyrosequencing Assay
3.2. Accuracy
3.3. Analytical Sensitivity
3.4. Intra- and Inter-Run Reproducibility
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Lumish, M.A.; Kohn, E.C.; Tew, W.P. Top advances of the year: Ovarian cancer. Cancer 2024, 130, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Durand, F.; Tang, R.; Pommier, M.; Nashvi, M.; Cotteret, S.; Genestie, C.; Le Formal, A.; Pautier, P.; Michels, J.; Kfoury, M.; et al. Clinical Relevance of BRCA1 Promoter Methylation Testing in Patients with Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 3124–3129. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Kalachand, R.D.; Stordal, B.; Madden, S.; Chandler, B.; Cunningham, J.; Goode, E.L.; Ruscito, I.; Braicu, E.I.; Sehouli, J.; Ignatov, A.; et al. BRCA1 Promoter Methylation and Clinical Outcomes in Ovarian Cancer: An Individual Patient Data Meta-Analysis. J. Natl. Cancer Inst. 2020, 112, 1190–1203. [Google Scholar] [CrossRef]
- Kawachi, A.; Yamashita, S.; Okochi-Takada, E.; Hirakawa, A.; Tsuda, H.; Shimomura, A.; Kojima, Y.; Yonemori, K.; Fujiwara, Y.; Kinoshita, T.; et al. BRCA1 promoter methylation in breast cancer patients is associated with response to olaparib/eribulin combination therapy. Breast Cancer Res. Treat. 2020, 181, 323–329. [Google Scholar] [CrossRef]
- Benhamida, J.K.; Hechtman, J.F.; Nafa, K.; Villafania, L.; Sadowska, J.; Wang, J.; Wong, D.; Zehir, A.; Zhang, L.; Bale, T.; et al. Reliable Clinical MLH1 Promoter Hypermethylation Assessment Using a High-Throughput Genome-Wide Methylation Array Platform. J. Mol. Diagn. 2020, 22, 368–375. [Google Scholar] [CrossRef]
- Higashimoto, K.; Hara, S.; Soejima, H. DNA Methylation Analysis Using Bisulfite Pyrosequencing. Methods Mol. Biol. 2023, 2577, 3–20. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barrière, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Krivak, T.C.; Kabil, N.; Munley, J.; Moore, K.N. PARP Inhibitors in Ovarian Cancer: A Review. Target Oncol. 2023, 18, 471–503. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Morganti, S.; Marra, A.; De Angelis, C.; Toss, A.; Licata, L.; Giugliano, F.; Taurelli Salimbeni, B.; Berton Giachetti, P.P.M.; Esposito, A.; Giordano, A.; et al. PARP Inhibitors for Breast Cancer Treatment: A Review. JAMA Oncol. 2024, 10, 658–670. [Google Scholar] [CrossRef]
- Taylor, A.K.; Kosoff, D.; Emamekhoo, H.; Lang, J.M.; Kyriakopoulos, C.E. PARP inhibitors in metastatic prostate cancer. Front. Oncol. 2023, 13, 1159557. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Saleem, M.A.; Mustafa, M.S. Promoter Hypermethylation of the BRCA1 Gene as a Novel Biomarker for Prostate Cancer. Cureus 2024, 16, e66467. [Google Scholar] [CrossRef]
- Zheng-Lin, B.; Rainone, M.; Varghese, A.M.; Yu, K.H.; Park, W.; Berger, M.; Mehine, M.; Chou, J.; Capanu, M.; Mandelker, D.; et al. Methylation Analyses Reveal Promoter Hypermethylation as a Rare Cause of “Second Hit” in Germline BRCA1-Associated Pancreatic Ductal Adenocarcinoma. Mol. Diagn. Ther. 2022, 26, 645–653. [Google Scholar] [CrossRef]
- Abdallah, R.; Zhao, S.; Garinet, S.; Hormigos, K.; Le Corre, D.; Cros, J.; Perez Toralla, K.; Bats, A.S.; Augustin, J.; Bachet, J.B.; et al. BRCA1 and RAD51C promotor methylation in human resectable pancreatic adenocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101880. [Google Scholar] [CrossRef]
- Sahnane, N.; Rivera, D.; Libera, L.; Carnevali, I.; Banelli, B.; Facchi, S.; Gismondi, V.; Paudice, M.; Cirmena, G.; Vellone, V.G.; et al. Pyrosequencing Assay for BRCA1 Methylation Analysis: Results from a Cross-Validation Study. J. Mol. Diagn. 2023, 25, 217–226. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | Pos. 7 | Pos. 8 | Pos. 9 | Pos. 10 | Pos. 11 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Mean | Pyro Result | TCGA Result | Real-Time PCR Result |
| 29 1694 | 55.2 | 71.3 | 53.6 | 49.9 | 54.1 | 53.1 | 58.6 | 53.9 | 48.5 | 43.8 | 43.3 | 53.2 | PRESENT | METH | |
| 29 1707 | 50.6 | 65.9 | 48.3 | 45.6 | 49.2 | 50.3 | 53.3 | 47.2 | 42.4 | 41.1 | 39.4 | 48.5 | PRESENT | METH | |
| 29 1711 | 61.9 | 71.8 | 58.7 | 55.9 | 58.7 | 58.5 | 65.4 | 61.2 | 58.8 | 52.5 | 53.6 | 59.7 | PRESENT | METH | |
| JB 2335 | 44.1 | 58.0 | 44.5 | 40.9 | 39.5 | 47.6 | 50.1 | 44.4 | 37.6 | 42.1 | 38.5 | 44.3 | PRESENT | METH | Positive |
| JB 2595 | 28.0 | 51.1 | 36.7 | 31.3 | 28.9 | 34.7 | 42.7 | 43.1 | 30.1 | 29.6 | 25.4 | 34.7 | PRESENT | METH | Positive |
| JB 2670 | 62.3 | 85.6 | 62.4 | 61.1 | 67.0 | 63.8 | 69.5 | 65.6 | 60.2 | 58.4 | 56.3 | 64.7 | PRESENT | METH | Positive |
| JB 1412 | 36.3 | 59.6 | 41.2 | 40.4 | 38.9 | 40.5 | 47.4 | 42.2 | 34.9 | 38.2 | 36.0 | 41.4 | PRESENT | Positive | |
| JB 1468 | 44.1 | 61.3 | 41.2 | 40.8 | 42.4 | 42.8 | 48.1 | 42.4 | 35.0 | 37.5 | 29.0 | 42.2 | PRESENT | Positive | |
| JB 3081 | 41.1 | 57.7 | 43.8 | 39.4 | 45.0 | 44.5 | 48.0 | 42.3 | 38.1 | 36.5 | 35.5 | 42.9 | PRESENT | Positive | |
| JB 3393 | 32.8 | 49.3 | 34.5 | 32.0 | 31.9 | 34.9 | 37.1 | 31.8 | 27.9 | 22.4 | 25.1 | 32.7 | PRESENT | Positive | |
| 29 1696 | 2.8 | 8.8 | 2.4 | 2.3 | 1.8 | 4.9 | 3.6 | 2.2 | 1.8 | 3.9 | 6.2 | 3.7 | Not Present | Not Meth | |
| 29 1710 | 2.5 | 9.8 | 2.5 | 3.1 | 2.4 | 5.8 | 3.9 | 2.1 | 3.1 | 3.9 | 6.2 | 4.1 | Not Present | Not Meth | |
| 29 1762 | 1.8 | 11.4 | 2.8 | 2.1 | 2.1 | 6.9 | 4.1 | 1.9 | 1.5 | 3.0 | 7.2 | 4.1 | Not Present | Not Meth | |
| 29 1763 | 3.8 | 24.6 | 7.3 | 6.4 | 3.7 | 11.8 | 8.8 | 5.2 | 4.6 | 8.9 | 11.5 | 8.8 | Not Present | Not Meth | |
| 29 1776 | 3.0 | 13.1 | 3.4 | 2.8 | 2.8 | 8.0 | 5.2 | 3.3 | 3.9 | 6.6 | 9.5 | 5.6 | Not Present | Not Meth | |
| 29 1784 | 2.2 | 9.7 | 3.8 | 2.9 | 2.5 | 6.5 | 4.6 | 2.4 | 2.0 | 4.0 | 7.3 | 4.4 | Not Present | Not Meth | |
| BRME 23 | 0.7 | 12.5 | 2.4 | 1.9 | 1.6 | 7.0 | 3.1 | 2.2 | 1.0 | 3.6 | 7.3 | 3.9 | Not Present | Negative | |
| JB 2206 | 1.8 | 10.8 | 1.7 | 1.5 | 1.5 | 4.5 | 2.9 | 1.2 | 1.4 | 2.9 | 5.3 | 3.2 | Not Present | Negative | |
| JB 3717 | 1.5 | 10.0 | 1.6 | 2.0 | 1.5 | 4.2 | 2.5 | 1.1 | 1.5 | 2.9 | 4.8 | 3.1 | Not Present | Negative | |
| JB 2388 | 2.1 | 9.5 | 3.0 | 3.0 | 2.2 | 5.4 | 4.2 | 2.5 | 1.9 | 4.5 | 6.5 | 4.1 | Not Present | Negative |
| (A) | |||||||||||||||
| Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | Pos. 7 | Pos. 8 | Pos. 9 | Pos. 10 | Pos. 11 | |||||
| Dilution | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Mean | |||
| Neg. | Run#1 | 1.4 | 14.5 | 3.3 | 3.2 | 2.1 | 7.0 | 5.1 | 2.4 | 2.2 | 5.5 | 8.9 | 5.1 | ||
| Neg. | Run#2 | 5.1 | 17.8 | 5.8 | 4.0 | 3.4 | 8.6 | 6.7 | 3.0 | 3.4 | 7.3 | 11.4 | 7.0 | ||
| 100% | Run#1 | 95.0 | 100.0 | 90.1 | 79.7 | 91.4 | 90.1 | 95.1 | 91.3 | 92.0 | 79.4 | 81.2 | 89.6 | ||
| 100% | Run#2 | 94.9 | 100.0 | 91.3 | 83.1 | 93.7 | 90.6 | 93.9 | 91.3 | 88.5 | 86.7 | 74.4 | 89.9 | ||
| 50% | Run#1 | 55.6 | 74.2 | 55.2 | 49.3 | 54.2 | 57.3 | 62.4 | 57.7 | 52.4 | 49.1 | 52.1 | 56.3 | ||
| 50% | Run#2 | 55.5 | 67.1 | 53.0 | 49.2 | 52.2 | 53.9 | 59.1 | 55.0 | 50.8 | 47.1 | 48.8 | 53.8 | ||
| 25% | Run#1 | 29.3 | 41.9 | 28.2 | 26.5 | 27.5 | 30.9 | 34.5 | 30.3 | 27.0 | 25.9 | 28.7 | 30.0 | ||
| 25% | Run#2 | 30.6 | 44.3 | 30.0 | 28.3 | 28.5 | 30.5 | 34.0 | 26.6 | 26.6 | 27.8 | 29.1 | 30.6 | ||
| 12.50% | Run#1 | 18.0 | 32.4 | 18.8 | 18.7 | 17.9 | 22.5 | 23.4 | 20.0 | 16.7 | 18.3 | 21.2 | 20.7 | ||
| 12.50% | Run#2 | 16.1 | 30.9 | 17.2 | 18.8 | 17.6 | 21.3 | 22.1 | 18.6 | 14.5 | 17.1 | 21.4 | 19.6 | ||
| 6.25% | Run#1 | 13.0 | 24.1 | 12.3 | 12.5 | 11.2 | 15.9 | 16.1 | 12.2 | 11.3 | 12.9 | 15.9 | 14.3 | ||
| 6.25% | Run#2 | 13.6 | 23.3 | 12.5 | 12.1 | 10.0 | 15.5 | 14.8 | 11.6 | 9.9 | 11.8 | 16.4 | 13.8 | ||
| 3.13% | Run#1 | 8.2 | 18.5 | 8.6 | 7.8 | 7.5 | 12.0 | 11.1 | 8.1 | 6.6 | 10.1 | 13.1 | 10.1 | ||
| 3.13% | Run#2 | 7.5 | 19.8 | 8.2 | 7.9 | 7.4 | 11.2 | 10.5 | 8.2 | 6.3 | 8.9 | 12.6 | 9.9 | ||
| 1.56% | Run#1 | 5.5 | 20.3 | 5.7 | 6.0 | 4.3 | 10.3 | 8.9 | 5.5 | 4.5 | 7.2 | 12.0 | 8.2 | ||
| 1.56% | Run#2 | 6.0 | 20.4 | 7.4 | 6.2 | 4.9 | 10.2 | 8.4 | 5.5 | 4.7 | 7.6 | 11.6 | 8.4 | ||
| (B) | |||||||||||||||
| Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | Pos. 7 | Pos. 8 | Pos. 9 | Pos. 10 | Pos. 11 | |||||
| Dilution | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Mean (measured) | Expected | Pyro Result | |
| 100% | Run#1 | 59.5 | 82.9 | 57.4 | 55.6 | 62.5 | 59.3 | 69.6 | 61.5 | 54.0 | 54.7 | 56.1 | 61.2 | DETECTED | |
| 100% | Run#2 | 72.0 | 87.1 | 70.2 | 61.0 | 71.8 | 71.0 | 80.5 | 67.8 | 65.3 | 54.2 | 61.4 | 69.3 | DETECTED | |
| 50% | Run#1 | 18.9 | 25.6 | 18.0 | 17.3 | 18.0 | 18.2 | 21.4 | 19.5 | 14.2 | 16.2 | 13.2 | 18.2 | 32.5 | DETECTED |
| 50% | Run#2 | 19.7 | 31.2 | 21.4 | 19.0 | 16.8 | 21.7 | 21.7 | 16.7 | 12.5 | 13.2 | 18.5 | 19.3 | 32.5 | DETECTED |
| 25% | Run#1 | 13.0 | 19.1 | 12.9 | 13.6 | 12.3 | 15.1 | 18.2 | 14.0 | 10.7 | 11.4 | 13.7 | 14.0 | 16.2 | DETECTED |
| 25% | Run#2 | 9.6 | 22.9 | 10.6 | 11.0 | 10.1 | 14.9 | 14.5 | 12.3 | 7.5 | 9.1 | 13.4 | 12.4 | 16.2 | DETECTED |
| 12.50% | Run#1 | 6.4 | 14.7 | 5.4 | 4.7 | 4.9 | 8.8 | 7.2 | 5.4 | 2.2 | 6.8 | 9.3 | 6.9 | 8.1 | NOT DETECTED |
| 12.50% | Run#2 | 7.7 | 16.9 | 8.6 | 7.5 | 6.7 | 11.2 | 12.1 | 7.9 | 5.0 | 8.9 | 12.9 | 9.6 | 8.1 | NOT DETECTED |
| 6.25% | Run#1 | 8.1 | 14.9 | 7.8 | 8.1 | 7.0 | 10.5 | 7.6 | 3.8 | 3.4 | 5.2 | 9.0 | 7.8 | 4.0 | NOT DETECTED |
| 6.25% | Run#2 | 6.2 | 16.9 | 7.3 | 8.7 | 4.9 | 11.6 | 9.9 | 5.9 | 4.4 | 8.2 | 10.9 | 8.6 | 4.0 | NOT DETECTED |
| 3.13% | Run#1 | 4.1 | 12.8 | 3.2 | 3.8 | 4.6 | 5.9 | 5.6 | 3.2 | 2.2 | 3.3 | 8.7 | 5.2 | 2.0 | NOT DETECTED |
| 3.13% | Run#2 | 3.3 | 11.4 | 5.4 | 3.6 | 3.5 | 6.2 | 4.1 | 2.6 | 2.6 | 4.4 | 6.0 | 4.8 | 2.0 | NOT DETECTED |
| (A) | |||||||||||||||
| Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | Pos. 7 | Pos. 8 | Pos. 9 | Pos. 10 | Pos. 11 | |||||
| Case # | ID | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Mean | Result | |
| 21 | JB 1412 | 50.6 | 84.4 | 49.9 | 48.3 | 49.7 | 54.4 | 53.9 | 51.8 | 40.8 | 44.1 | 45.8 | 52.2 | PRESENT | |
| 21 | JB 1412 | 36.3 | 59.6 | 41.2 | 40.4 | 38.9 | 40.5 | 47.4 | 42.2 | 34.9 | 38.2 | 36.0 | 41.4 | PRESENT | |
| 21 | JB1412 | 50.9 | 76.8 | 47.5 | 44.3 | 46.1 | 49.1 | 55.4 | 49.8 | 45.6 | 44.7 | 45.6 | 50.5 | PRESENT | |
| 22 | JB 1468 | 41.9 | 63.4 | 39.6 | 39.4 | 40.6 | 41.3 | 47.4 | 41.7 | 35.7 | 37.4 | 28.5 | 41.5 | PRESENT | |
| 22 | JB 1468 | 42.0 | 62.0 | 41.0 | 38.5 | 39.5 | 40.7 | 46.7 | 41.7 | 36.2 | 36.2 | 29.4 | 41.3 | PRESENT | |
| 22 | JB 1468 | 43.0 | 60.4 | 41.6 | 40.9 | 42.5 | 41.4 | 47.8 | 42.2 | 35.6 | 39.0 | 29.9 | 42.2 | PRESENT | |
| 23 | JB 2400 | 3.0 | 13.1 | 3.3 | 2.5 | 1.6 | 6.5 | 4.3 | 1.8 | 2.4 | 4.2 | 7.3 | 4.5 | Not Present | |
| 23 | JB 2400 | 3.4 | 12.0 | 4.2 | 3.2 | 2.5 | 7.6 | 4.9 | 2.6 | 1.7 | 4.9 | 6.7 | 4.9 | Not Present | |
| 23 | JB 2400 | 2.7 | 14.0 | 4.0 | 3.5 | 1.7 | 6.8 | 4.5 | 2.3 | 1.9 | 4.5 | 7.2 | 4.8 | Not Present | |
| 24 | JB 2505 | 61.1 | 97.5 | 66.3 | 63.1 | 67.4 | 68.2 | 75.0 | 67.2 | 62.4 | 61.9 | 61.0 | 68.3 | PRESENT | |
| 24 | JB 2505 | 65.4 | 90.3 | 65.3 | 60.8 | 66.3 | 67.1 | 72.4 | 67.7 | 61.5 | 60.8 | 60.0 | 67.0 | PRESENT | |
| 24 | JB 2505 | 62.6 | 89.7 | 66.0 | 58.2 | 63.6 | 66.3 | 71.2 | 64.3 | 63.6 | 60.3 | 59.0 | 65.9 | PRESENT | |
| 25 | JB 2520 | 4.0 | 13.7 | 5.0 | 3.6 | 2.7 | 7.0 | 5.5 | 3.0 | 2.5 | 5.9 | 8.7 | 5.6 | Not Present | |
| 25 | JB 2520 | 2.2 | 14.5 | 3.6 | 2.9 | 2.4 | 6.9 | 4.4 | 3.1 | 2.8 | 4.6 | 9.0 | 5.1 | Not Present | |
| 25 | JB 2520 | 2.7 | 18.0 | 3.4 | 2.2 | 2.2 | 6.0 | 7.3 | 2.9 | 2.4 | 5.1 | 8.6 | 5.5 | Not Present | |
| 26 | JB 2540 | 2.7 | 12.8 | 4.0 | 4.2 | 3.9 | 7.5 | 5.9 | 3.2 | 2.1 | 4.7 | 7.0 | 5.3 | Not Present | |
| 26 | JB 2540 | 5.8 | 12.3 | 5.1 | 4.9 | 3.2 | 8.5 | 5.6 | 2.7 | 2.3 | 5.6 | 9.6 | 6.0 | Not Present | |
| 26 | JB 2540 | 3.7 | 15.0 | 5.4 | 5.1 | 4.6 | 7.7 | 5.7 | 3.5 | 3.2 | 5.9 | 6.5 | 6.0 | Not Present | |
| (B) | |||||||||||||||
| Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | Pos. 7 | Pos. 8 | Pos. 9 | Pos. 10 | Pos. 11 | |||||
| Case # | ID | Run# | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Meth. (%) | Mean | Result |
| 16 | BRME 23 | Run#1 | 0.7 | 12.5 | 2.4 | 1.9 | 1.6 | 7.0 | 3.1 | 2.2 | 1.0 | 3.6 | 7.3 | 3.9 | Not Present |
| 16 | BRME 23 | Run#2 | 2.7 | 11.0 | 2.9 | 3.1 | 0.8 | 8.3 | 2.0 | 0.0 | 1.5 | 9.6 | 9.6 | 4.7 | Not Present |
| 16 | BRME 23 | Run#3 | 3.4 | 17.3 | 3.9 | 4.5 | 5.0 | 9.1 | 7.3 | 4.1 | 3.4 | 6.9 | 8.0 | 6.6 | Not Present |
| 17 | JB 1468 | Run#1 | 42.0 | 62.0 | 41.0 | 38.5 | 39.5 | 40.7 | 46.7 | 41.7 | 36.2 | 36.2 | 29.4 | 41.3 | PRESENT |
| 17 | JB 1468 | Run#2 | 28.7 | 56.0 | 36.9 | 35.4 | 36.1 | 36.7 | 37.1 | 38.5 | 34.2 | 33.9 | 27.6 | 36.5 | PRESENT |
| 17 | JB 1468 | Run#3 | 34.3 | 62.1 | 40.2 | 38.7 | 40.3 | 42.0 | 46.4 | 41.3 | 33.2 | 36.7 | 30.5 | 40.5 | PRESENT |
| 13 | JB 2335 | Run#1 | 48.3 | 63.6 | 46.7 | 44.6 | 44.6 | 41.6 | 55.5 | 41.3 | 31.6 | 35.5 | 36.2 | 44.5 | PRESENT |
| 13 | JB 2335 | Run#2 | 44.1 | 58.0 | 44.5 | 40.9 | 39.5 | 47.6 | 50.1 | 44.4 | 37.6 | 42.1 | 38.5 | 44.3 | PRESENT |
| 13 | JB 2335 | Run#3 | 38.4 | 73.0 | 47.7 | 41.2 | 40.8 | 48.3 | 52.8 | 44.5 | 35.4 | 34.6 | 42.0 | 45.3 | PRESENT |
| 18 | JB 2400 | Run#1 | 3.4 | 12.0 | 4.2 | 3.2 | 2.5 | 7.6 | 4.9 | 2.6 | 1.7 | 4.9 | 6.7 | 4.9 | Not Present |
| 18 | JB 2400 | Run#2 | 2.5 | 12.1 | 3.8 | 4.1 | 3.9 | 6.4 | 0.0 | 0.0 | 0.0 | 8.0 | 4.8 | 4.1 | Not Present |
| 18 | JB 2400 | Run#3 | 3.3 | 8.8 | 3.5 | 2.9 | 3.1 | 6.0 | 5.0 | 4.9 | 3.1 | 4.2 | 4.8 | 4.5 | Not Present |
| 19 | JB 2540 | Run#1 | 4.9 | 16.9 | 7.0 | 6.9 | 3.7 | 8.2 | 8.5 | 4.2 | 4.8 | 6.6 | 11.8 | 7.6 | Not Present |
| 19 | JB 2540 | Run#2 | 2.3 | 10.6 | 2.8 | 2.8 | 3.2 | 5.3 | 7.4 | 3.2 | 2.8 | 3.7 | 7.5 | 4.7 | Not Present |
| 19 | JB 2540 | Run#3 | 4.3 | 28.5 | 8.2 | 7.6 | 5.3 | 12.5 | 13.0 | 6.2 | 3.9 | 7.3 | 11.1 | 9.8 | Not Present |
| 20 | JB 3393 | Run#1 | 32.8 | 49.3 | 34.5 | 32.0 | 31.9 | 34.9 | 37.1 | 31.8 | 27.9 | 22.4 | 25.1 | 32.7 | PRESENT |
| 20 | JB 3393 | Run#2 | 32.3 | 44.7 | 33.0 | 33.3 | 33.1 | 33.0 | 39.1 | 33.9 | 28.9 | 24.6 | 22.1 | 32.5 | PRESENT |
| 20 | JB 3393 | Run#3 | 23.6 | 59.9 | 32.2 | 31.1 | 29.6 | 35.3 | 37.9 | 31.6 | 24.9 | 24.8 | 26.4 | 32.5 | PRESENT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacares, R.; Soslow, R.; Olvera, N.; Levine, D.A.; Zhang, L. A Rapid and Reliable Test for BRCA1 Promoter Hypermethylation in Paraffin Tissue Using Pyrosequencing. Diagnostics 2025, 15, 601. https://doi.org/10.3390/diagnostics15050601
Bacares R, Soslow R, Olvera N, Levine DA, Zhang L. A Rapid and Reliable Test for BRCA1 Promoter Hypermethylation in Paraffin Tissue Using Pyrosequencing. Diagnostics. 2025; 15(5):601. https://doi.org/10.3390/diagnostics15050601
Chicago/Turabian StyleBacares, Ruben, Robert Soslow, Narciso Olvera, Douglas A. Levine, and Liying Zhang. 2025. "A Rapid and Reliable Test for BRCA1 Promoter Hypermethylation in Paraffin Tissue Using Pyrosequencing" Diagnostics 15, no. 5: 601. https://doi.org/10.3390/diagnostics15050601
APA StyleBacares, R., Soslow, R., Olvera, N., Levine, D. A., & Zhang, L. (2025). A Rapid and Reliable Test for BRCA1 Promoter Hypermethylation in Paraffin Tissue Using Pyrosequencing. Diagnostics, 15(5), 601. https://doi.org/10.3390/diagnostics15050601