
Integrated Meta-Analysis Identifies Keratin Family Genes and Associated Genes as Key Biomarkers and Therapeutic Targets in Metastatic Cutaneous Melanoma


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Selection and Preprocessing (Inclusion-Exclusion Criteria)
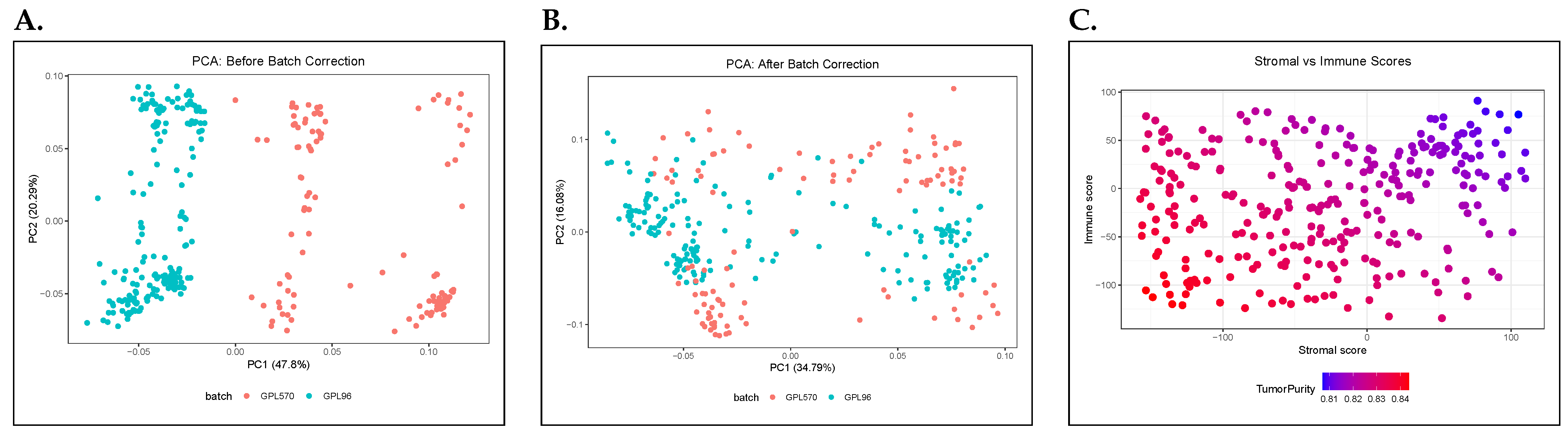
2.2. Quality Control (QC) and Batch-Effect Correction
2.3. Tumor Purity Filtering
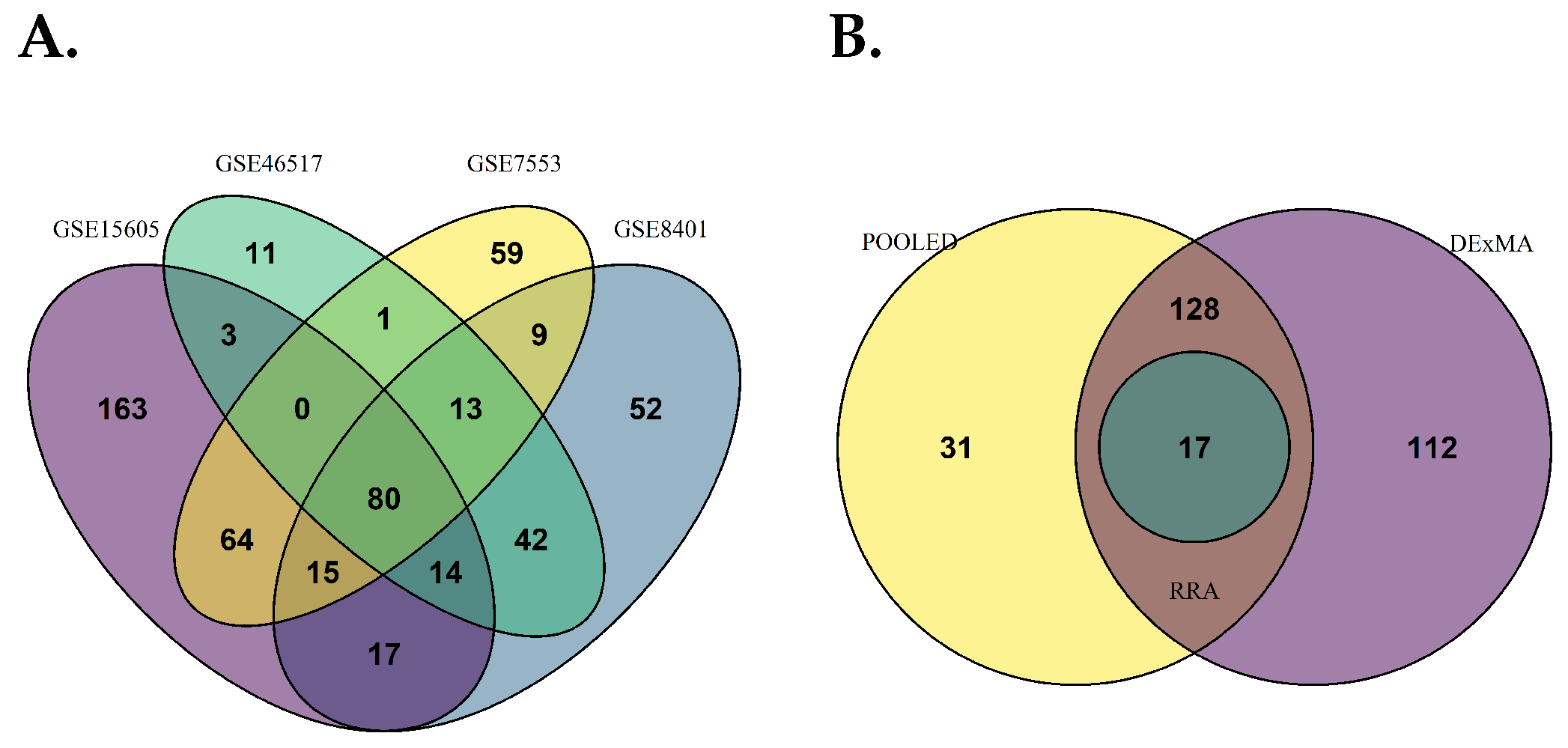
2.4. Integrated Meta-Analysis of Differentially Expressed Genes
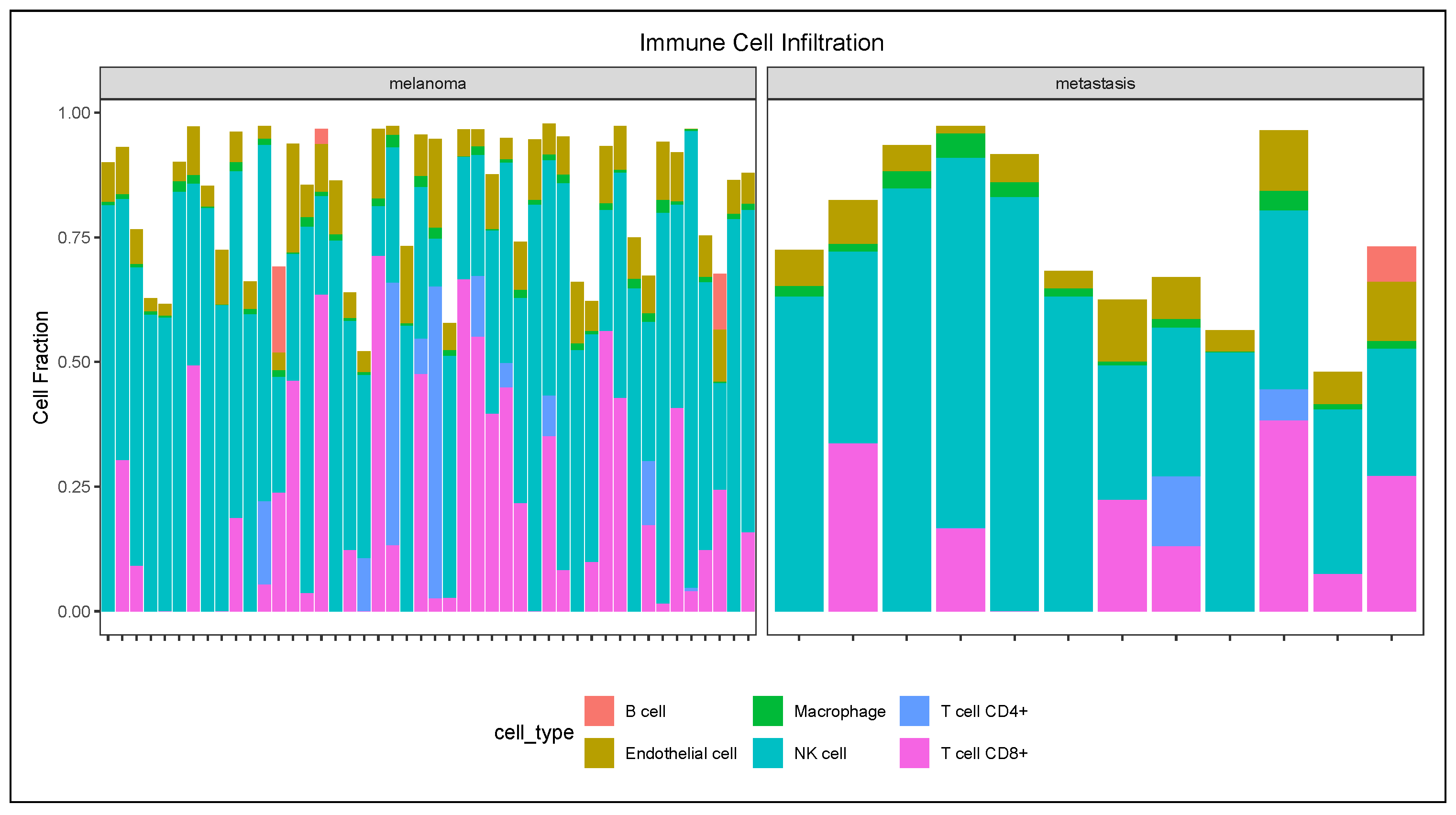
2.5. Immune Cell Infiltration Analysis
2.6. Functional Enrichment Analysis of DEGs
2.7. Protein-Protein Interaction Network and Hub Gene Identification
2.8. Validation and Prognostic Assessment of Hub Genes (TCGA-SKCM)
2.9. Drug–Gene Interaction and Structural Druggability Analysis
3. Results
3.1. Data Selection, Quality Control, and Tumor Purity Filtering
3.2. Integrated Meta-Analysis of Differentially Expressed Genes
3.3. Immune Cell Infiltration Analysis
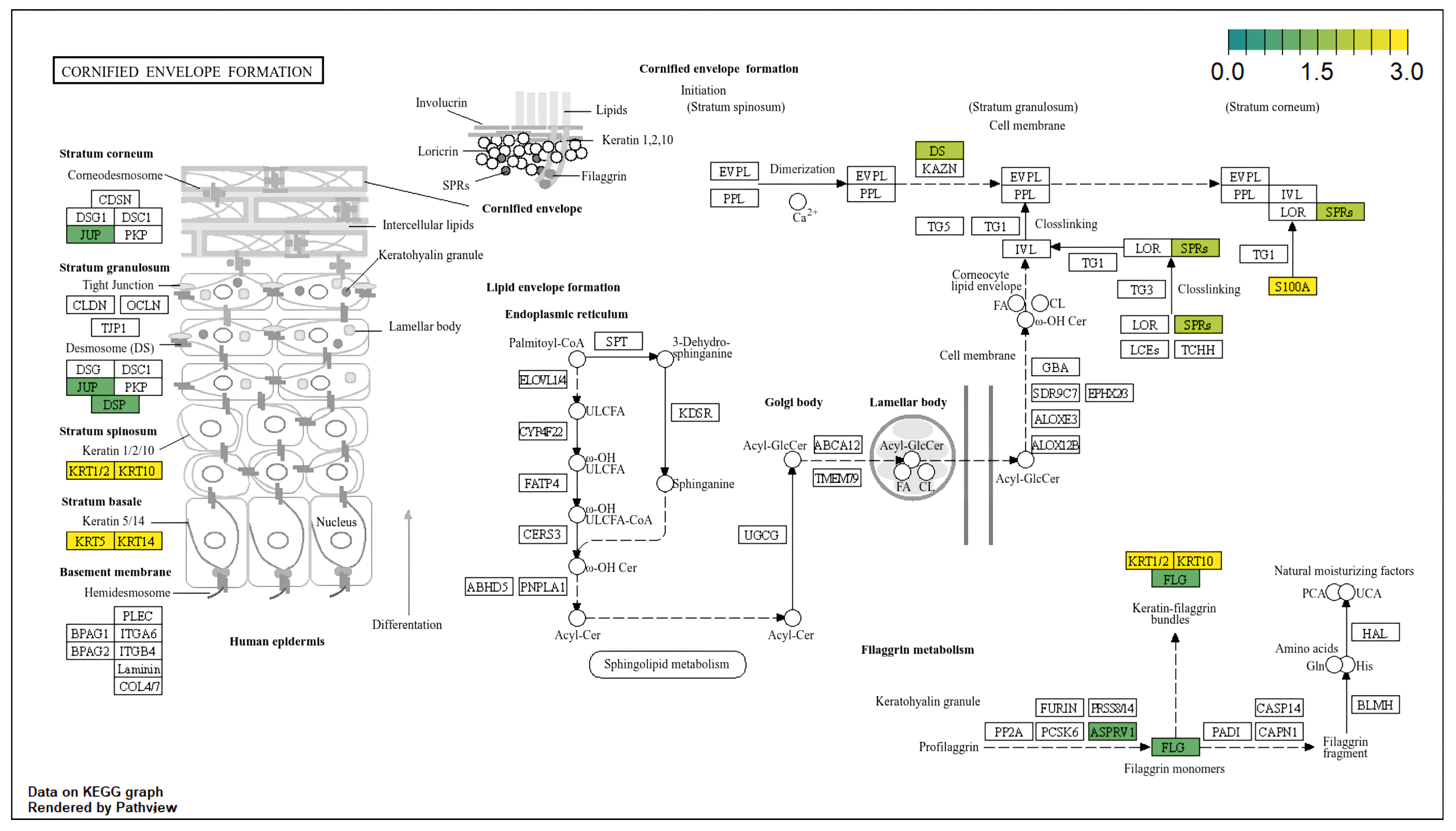
3.4. Functional Enrichment Analysis (KEGG/GO)
3.5. Protein-Protein Network and Hub Gene Identification
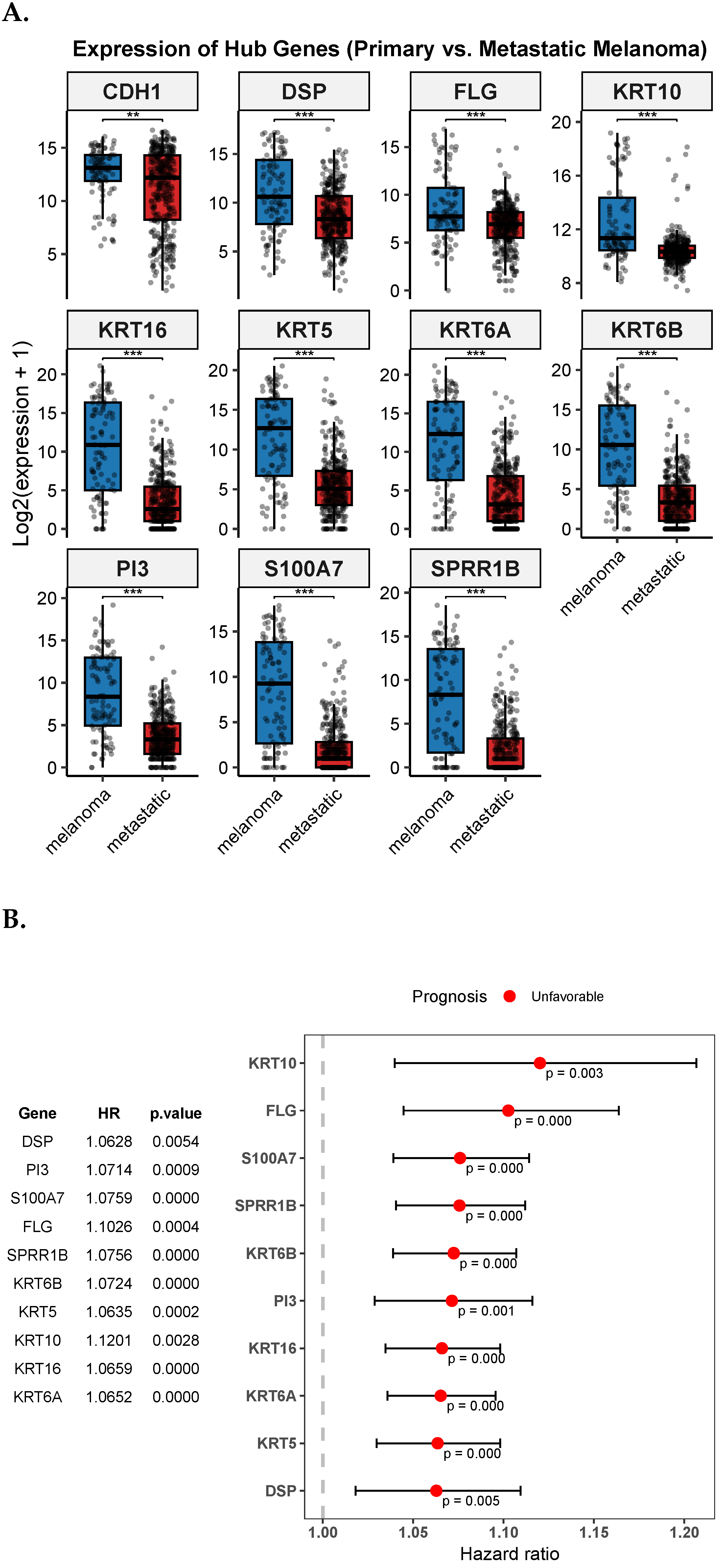
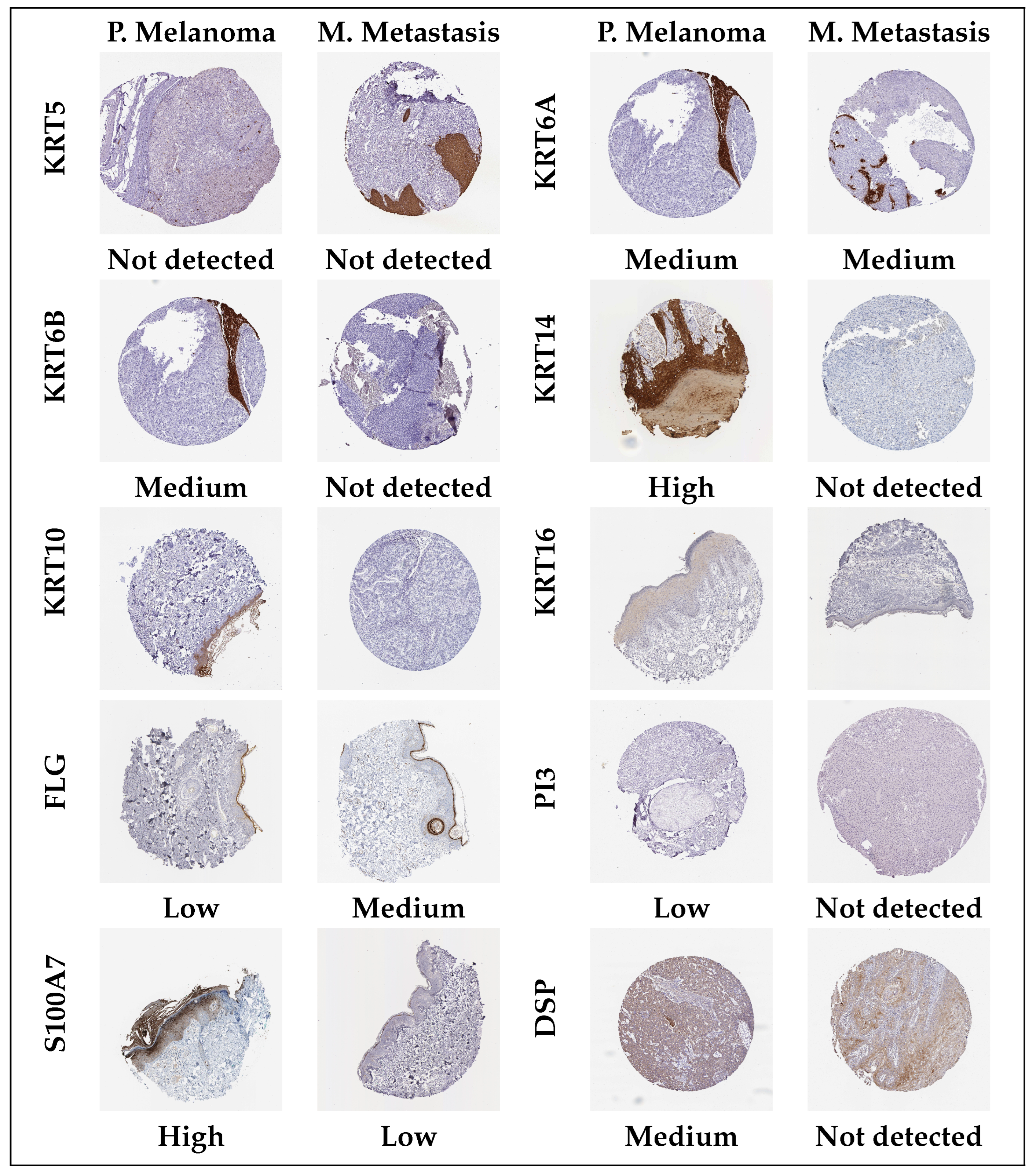
3.6. Validation and Prognostic Assessment of Hub Genes (TCGA-SKCM)
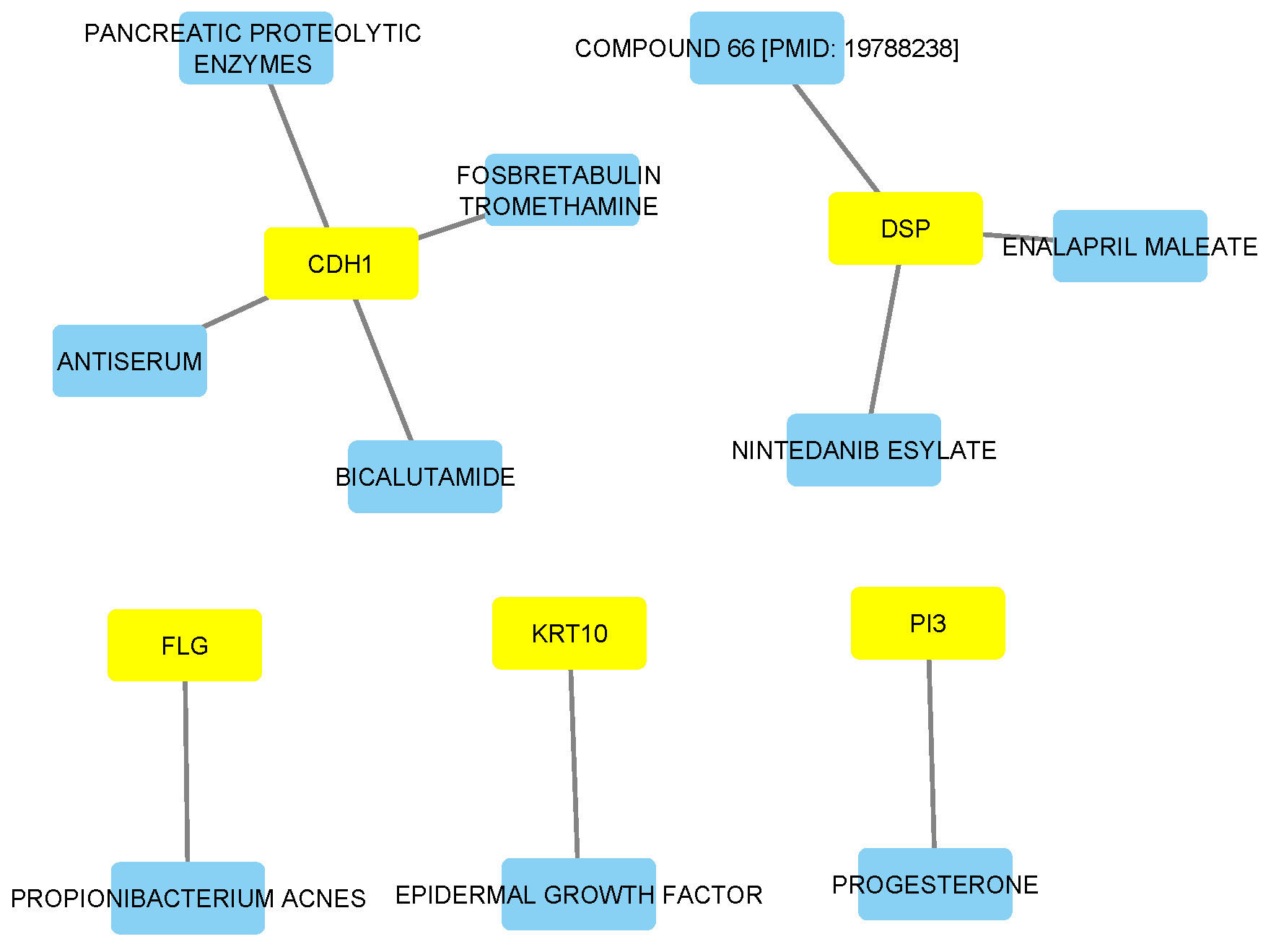
3.7. Drug-Gene Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Yang, R.; Wang, Z.; Li, J.; Pi, X.; Gao, R.; Ma, J.; Qing, Y.; Zhou, S. The identification of the metabolism subtypes of skin cutaneous melanoma associated with the tumor microenvironment and the immunotherapy. Front. Cell Dev. Biol. 2021, 9, 707677. [Google Scholar] [CrossRef]
- Key Statistics for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/types/melanoma-skin-cancer/about/key-statistics.html (accessed on 24 April 2025).
- Key Statistics for Melanoma Skin Cancer. Risk of Getting Melanoma. 2025. Available online: https://www.cancer.org/cancer/types/melanoma-skin-cancer/causes-risks-prevention/risk-factors.html (accessed on 24 April 2025).
- Durgham, R.A.; Nassar, S.I.; Gun, R.; Nguyen, S.A.; Asarkar, A.A.; Nathan, C.A.O. The Prognostic Value of the 31-Gene Expression Profile Test in Cutaneous Melanoma: A Systematic Review and Meta-Analysis. Cancers 2024, 16, 3714. [Google Scholar] [CrossRef]
- Sharif-Askari, N.S.; Sharif-Askari, F.S.; Guraya, S.Y.; Bendardaf, R.; Hamoudi, R. Integrative systematic review meta-analysis and bioinformatics identifies MicroRNA-21 and its target genes as biomarkers for colorectal adenocarcinoma. Int. J. Surg. 2020, 73, 113–122. [Google Scholar] [CrossRef]
- Atkins, M.B.; Curiel-Lewandrowski, C.; Fisher, D.E.; Swetter, S.M.; Tsao, H.; Aguirre-Ghiso, J.A.; Soengas, M.S.; Weeraratna, A.T.; Flaherty, K.T.; Herlyn, M.; et al. The state of melanoma: Emergent challenges and opportunities. Clin. Cancer Res. 2021, 27, 2678–2697. [Google Scholar] [CrossRef]
- Ali, M.D.; Mazhar, T.; Shahzad, T.; Rehman, W.U.; Shahid, M.; Hamam, H. An Advanced Deep Learning Framework for Skin Cancer Classification. Rev. Socionetwork Strateg. 2025, 19, 111–130. [Google Scholar] [CrossRef]
- Khan, M.A.; Mazhar, T.; Ali, M.D.; Khattak, U.F.; Shahzad, T.; Saeed, M.M.; Hamam, H. Automatic melanoma and non-melanoma skin cancer diagnosis using advanced adaptive fine-tuned convolution neural networks. Discov. Oncol. 2025, 16, 645. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Das, A.; Ghose, A.; Naicker, K.; Sanchez, E.; Chargari, C.; Rassy, E.; Boussios, S. Advances in adoptive T-cell therapy for metastatic melanoma. Curr. Res. Transl. Med. 2023, 71, 103404. [Google Scholar] [CrossRef]
- Hu, Q.; Xuan, J.; Wang, L.; Shen, K.; Gao, Z.; Zhou, Y.; Wei, C.; Gu, J. Application of adoptive cell therapy in malignant melanoma. J. Transl. Med. 2025, 23, 102. [Google Scholar] [CrossRef]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.P.; Lijnsvelt, J.M.; Maher, N.G.; et al. Neoadjuvant nivolumab and ipilimumab in resectable stage III melanoma. N. Engl. J. Med. 2024, 391, 1696–1708. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Steininger, J.; Gellrich, F.F.; Schulz, A.; Westphal, D.; Beissert, S.; Meier, F. Systemic therapy of metastatic melanoma: On the road to cure. Cancers 2021, 13, 1430. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Al Sharie, A.H.; Al Zu’bi, Y.O.; El-Elimat, T.; Al-Kammash, K.; Abu Lil, A.; Isawi, I.H.; Al Sharie, S.; Abu Mousa, B.M.; Al Malkawi, A.A.; Alali, F.Q. ANO4 expression is a potential prognostic biomarker in non-metastasized clear cell renal cell carcinoma. J. Pers. Med. 2023, 13, 295. [Google Scholar] [CrossRef]
- Gupta, S.; Gupta, M.K.; Shabaz, M.; Sharma, A. Deep learning techniques for cancer classification using microarray gene expression data. Front. Physiol. 2022, 13, 952709. [Google Scholar] [CrossRef]
- Li, K.; Guo, S.; Tong, S.; Sun, Q.; Jin, S.; Qi, B.; Shao, Y.; Xu, N. Identification of Keratinocyte Differentiation-Involved Genes for Metastatic Melanoma by Gene Expression Profiles. Comput. Math. Methods Med. 2021, 2021, 9652768. [Google Scholar] [CrossRef]
- Luan, H.; Jian, L.; He, Y.; Zhang, T.; Zhou, L. Exploration and validation of metastasis-associated genes for skin cutaneous melanoma. Sci. Rep. 2022, 12, 13002. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the Topology of Active Sites: On the Prediction of Pockets and Subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [Google Scholar] [CrossRef]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 1–16. [Google Scholar] [CrossRef]
- Raskin, L.; Fullen, D.R.; Giordano, T.J.; Thomas, D.G.; Frohm, M.L.; Cha, K.B.; Ahn, J.; Mukherjee, B.; Johnson, T.M.; Gruber, S.B. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Investig. Dermatol. 2013, 133, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Kabbarah, O.; Nogueira, C.; Feng, B.; Nazarian, R.M.; Bosenberg, M.; Wu, M.; Scott, K.L.; Kwong, L.N.; Xiao, Y.; Cordon-Cardo, C.; et al. Integrative genome comparison of primary and metastatic melanomas. PLoS ONE 2010, 5, e10770. [Google Scholar] [CrossRef]
- Jalili, A.; Mertz, K.D.; Romanov, J.; Wagner, C.; Kalthoff, F.; Stuetz, A.; Pathria, G.; Gschaider, M.; Stingl, G.; Wagner, S.N. NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo. PLoS ONE 2013, 8, e69064. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Villatoro-García, J.A.; Martorell-Marugán, J.; Toro-Domínguez, D.; Román-Montoya, Y.; Femia, P.; Carmona-Sáez, P. DExMA: An R package for performing gene expression meta-analysis with missing genes. Mathematics 2022, 10, 3376. [Google Scholar] [CrossRef]
- Cui, H.; Zhao, G.; Lu, Y.; Zuo, S.; Duan, D.; Luo, X.; Zhao, H.; Li, J.; Zeng, Z.; Chen, Q.; et al. TIMER3: An enhanced resource for tumor immune analysis. Nucleic Acids Res. 2025, 53, W534–W541. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2017, 46, D1068–D1073. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Redoute-Timonnier, C.; Auguste, P. Implication of the extracellular matrix in metastatic tumor cell dormancy. Cancers 2024, 16, 4076. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Li, B.; Jia, L.; Li, Y. Identification of core genes and pathways in melanoma metastasis via bioinformatics analysis. Int. J. Mol. Sci. 2022, 23, 794. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Czyz, M. WNT signaling in melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Wessely, A.; Steeb, T.; Berking, C.; Heppt, M.V. How neural crest transcription factors contribute to melanoma heterogeneity, cellular plasticity, and treatment resistance. Int. J. Mol. Sci. 2021, 22, 5761. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ribero, S.; Cucci, M.A.; Grattarola, M.; Monge, C.; Dianzani, C.; Barrera, G.; Muzio, G. Oxidative stress-related mechanisms in melanoma and in the acquired resistance to targeted therapies. Antioxidants 2021, 10, 1942. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Context-dependent roles of claudins in tumorigenesis. Front. Oncol. 2021, 11, 676781. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, M.; Gao, T.; Xu, X.; Yu, W.; Liu, J.; Liu, R.; Zhang, X.; Li, F.; Hai, C. Identification of key genes linking bisphenols exposure and breast cancer. Toxicology 2025, 514, 154123. [Google Scholar] [CrossRef]
- Hao, X.; Han, F.; Ma, B.; Zhang, N.; Chen, H.; Jiang, X.; Yin, L.; Liu, W.; Ao, L.; Cao, J.; et al. SOX30 is a key regulator of desmosomal gene suppressing tumor growth and metastasis in lung adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 111. [Google Scholar] [CrossRef]
- Harada, K.; Sakamoto, N.; Kitaoka, T.; Nakamura, Y.; Kondo, R.; Morisue, R.; Hashimoto, H.; Yamamoto, Y.; Ukai, S.; Maruyama, R.; et al. PI3 expression predicts recurrence after chemotherapy with DNA-damaging drugs in gastric cancer. J. Pathol. 2025, 265, 472–485. [Google Scholar] [CrossRef]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.L.; Yang, F.; Li, R.N. Identification of potential Core genes between primary and metastatic malignant melanoma and analysis of their immune correlation. Int. J. Gen. Med. 2022, 15, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Iams, W.T.; Sosman, J.A.; Chandra, S. Novel targeted therapies for metastatic melanoma. Cancer J. 2017, 23, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Hoffner, B.; Benchich, K. Trametinib: A targeted therapy in metastatic melanoma. J. Adv. Pract. Oncol. 2018, 9, 741. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Melanoma | |||
|---|---|---|---|
| GEO Name | Platform | Primary | Metastatic |
| GSE7553 [23] | GPL570 (Affymetrix Human Genome U133 Plus 2.0) | 46 | 12 |
| GSE15605 [24] | GPL570 (Affymetrix Human Genome U133 Plus 2.0) | 31 | 52 |
| GSE46517 [25] | GPL96 (Affymetrix Human Genome U133A) | 31 | 73 |
| GSE8401 [26] | GPL96 (Affymetrix Human Genome U133A) | 14 | 40 |
| Gene | Drug | Approval Status | Indication | Interaction Score |
|---|---|---|---|---|
| PI3 | PROGESTERONE | Approved | Pre-term birth risk reduction | 1.77 |
| KRT10 | EPIDERMAL GROWTH FACTOR | Not Approved | Not available | 11.60 |
| DSP | COMPOUND 66 [PMID: 19788238] | Not Approved | Not available | 1.58 |
| DSP | ENALAPRIL MALEATE | Approved | Antihypertensive agent | 1.83 |
| DSP | NINTEDANIB ESYLATE | Approved | Antineoplastic agent | 1.39 |
| CDH1 | FOSBRETABULIN TROMETHAMINE | Not Approved | Not available | 13.05 |
| CDH1 | ANTISERUM | Not Approved | Not available | 0.75 |
| CDH1 | BICALUTAMIDE | Approved | Antineoplastic agent | 1.31 |
| CDH1 | PANCREATIC PROTEOLYTIC ENZYMES | Not Approved | Not available | 6.53 |
| FLG | PROPIONIBACTERIUM ACNES | Not Approved | Not available | 52.20 |
| Pockets | Gene | Druggability_Score | Volume |
|---|---|---|---|
| P 0 | CDH1 | 0.80 | 1903.56 |
| P 0 | DSP | 0.82 | 792.39 |
| P 0 | FLG | 0.80 | 3880.47 |
| P 0 | PI3 | 0.84 | 760.04 |
| P 0 | S100A7 | 0.76 | 615.68 |
| P 0 | SPRR1B | 0.26 | 109.65 |
| P 0 | KRT5 | 0.58 | 113.20 |
| P 0 | KRT6A | 0.71 | 374.28 |
| P 0 | KRT6B | 0.78 | 1058.92 |
| P 0 | KRT10 | 0.60 | 396.39 |
| P 0 | KRT16 | 0.80 | 1059.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abubakari, S.; Dizman, Y.A.; Karaman, F. Integrated Meta-Analysis Identifies Keratin Family Genes and Associated Genes as Key Biomarkers and Therapeutic Targets in Metastatic Cutaneous Melanoma. Diagnostics 2025, 15, 1770. https://doi.org/10.3390/diagnostics15141770
Abubakari S, Dizman YA, Karaman F. Integrated Meta-Analysis Identifies Keratin Family Genes and Associated Genes as Key Biomarkers and Therapeutic Targets in Metastatic Cutaneous Melanoma. Diagnostics. 2025; 15(14):1770. https://doi.org/10.3390/diagnostics15141770
Chicago/Turabian StyleAbubakari, Sumaila, Yeşim Aktürk Dizman, and Filiz Karaman. 2025. "Integrated Meta-Analysis Identifies Keratin Family Genes and Associated Genes as Key Biomarkers and Therapeutic Targets in Metastatic Cutaneous Melanoma" Diagnostics 15, no. 14: 1770. https://doi.org/10.3390/diagnostics15141770
APA StyleAbubakari, S., Dizman, Y. A., & Karaman, F. (2025). Integrated Meta-Analysis Identifies Keratin Family Genes and Associated Genes as Key Biomarkers and Therapeutic Targets in Metastatic Cutaneous Melanoma. Diagnostics, 15(14), 1770. https://doi.org/10.3390/diagnostics15141770