
Fibrotic Hypersensitivity Pneumonitis: A Diagnostic Challenge Leading to Lung Transplantation

 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HP | Hypersensitivity Pneumonitis |
| HRCT | High-Resolution Computed Tomography |
| ILD | Interstitial Lung Disease |
| IPF | Idiopathic Pulmonary Fibrosis |
| PFT | Pulmonary Function Test |
| FVC | Forced Vital Capacity |
| DLCO | Diffusing Capacity of the Lung for Carbon Monoxide |
| BAL | Bronchoalveolar Lavage |
| EKG | Electrocardiogram |
| ANA | Anti-Nuclear-Antigen antibody panel |
| IgG | Immunoglobulin G |
| FEV | Forced Expiratory Volume |
| TLC | Total Lung Capacity |
| RV | Residual Volume |
| IgE | Immunoglobulin E |
| COPD | Chronic Obstructive Pulmonary Disease |
| ERV | Expiratory Reserve Volume |
| RB-ILD | Respiratory Bronchiolitis-Associated Interstitial Lung Disease |
| PaO2 | Partial Pressure of Oxygen |
| HE | Hematoxylin and Eosin |
| PaCO2 | Partial Pressure of Carbon Dioxide |
| PPF | Progressive Pulmonary Fibrosis |
| ABG | Arterial Blood Gas |
| MDD | Multidisciplinary discussion |
| ATS | American Thoracic Society |
| JRS | Japanese Respiratory Society |
| ALAT | Latin American Thoracic Association |
| Anti-nRNP/Sm | Anti-nuclear Ribonucleoprotein/Smith antibodies |
| Anti-Sm | Anti-Smith antibodies |
| Anti-SS-A (Ro) | Anti-SS-A (Ro) antibodies |
| Anti-Ro-52 | Anti-Ro-52 antibodies |
| Anti-SS-B (La) | Anti-SS-B (La) antibodies |
| Anti-Scl-70 | Anti-topoisomerase I (Scl-70) antibodies |
| Anti-PM-Scl 100 | Anti-PM-Scl 100 antibodies |
| Anti-Jo-1 | Anti-histidyl-tRNA synthetase (Jo-1) antibodies |
| Anti-Centromere B | Anti-centromere B antibodies |
| Anti-PCNA | Anti-proliferating cell nuclear antigen (PCNA) antibodies |
| Anti-dsDNA | Anti-double-stranded DNA antibodies |
| Anti-Nucleosome | Anti-nucleosome antibodies |
| Anti-Histone | Anti-histone antibodies |
| Anti-Ribosomal P Protein | Anti-ribosomal P protein antibodies |
| Anti-AMA-M2 | Anti-mitochondrial antibodies type M2 |
| Anti-DFS70 | Anti-dense fine speckled 70 antibodies |
| DIP | Desquamative Interstitial Pneumonia |
| SaO2 | Arterial Oxygen Saturation |
| HCO3− | Bicarbonate |
| MEF25 | Maximal Expiratory Flow at 25% of Forced Vital Capacity |
References
- Dabiri, M.; Jehangir, M.; Khoshpouri, P.; Chalian, H. Hypersensitivity Pneumonitis: A Pictorial Review Based on the New ATS/JRS/ALAT Clinical Practice Guideline for Radiologists and Pulmonologists. Diagnostics 2022, 12, 2874. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Paul, I. Insights on immune profile, pathogenesis and differential diagnosis of hypersensitivity pneumonitis and pulmonary sarcoidosis–A holistic review and bibliometric analysis. Respir. Investig. 2025, 63, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Vasakova, M.; Morell, F.; Walsh, S.; Leslie, K.; Raghu, G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2017, 196, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Montero-Fernandez, M.A.; Kern, I.; Pezzuto, F.; Lunardi, F.; Hofman, P.; Berezowska, S.; Attanoos, R.; Burke, L.; Mason, P.; et al. The role of pathologists in the diagnosis of occupational lung diseases: An expert opinion of the European Society of Pathology Pulmonary Pathology Working Group. Virchows Arch. 2024, 485, 173–195. [Google Scholar] [CrossRef]
- Calaras, D.; David, A.; Vasarmidi, E.; Antoniou, K.; Corlateanu, A. Hypersensitivity Pneumonitis: Challenges of a Complex Disease. Can. Respir. J. 2024, 2024, 4919951. [Google Scholar] [CrossRef]
- Oliveira, M.; Oliveira, D.; Lisboa, C.; Boechat, J.L.; Delgado, L. Clinical Manifestations of Human Exposure to Fungi. J. Fungi 2023, 9, 381. [Google Scholar] [CrossRef]
- Salisbury, M.L.; Myers, J.L.; Belloli, E.A.; Kazerooni, E.A.; Martinez, F.J.; Flaherty, K.R. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar] [CrossRef]
- Petnak, T.; Thongprayoon, C.; Baqir, M.; Ryu, J.H.; Moua, T. Antigen identification and avoidance on outcomes in fibrotic hypersensitivity pneumonitis. Eur. Respir. J. 2022, 60, 2101336. [Google Scholar] [CrossRef]
- Sanduzzi Zamparelli, S.; Sanduzzi Zamparelli, A.; Bocchino, M. The Evolving Concept of the Multidisciplinary Approach in the Diagnosis and Management of Interstitial Lung Diseases. Diagnostics 2023, 13, 2437. [Google Scholar] [CrossRef]
- Bergantini, L.; Nardelli, G.; d’Alessandro, M.; Montuori, G.; Piccioli, C.; Rosi, E.; Gangi, S.; Cavallaro, D.; Cameli, P.; Bargagli, E. Combined Sarcoidosis and Idiopathic Pulmonary Fibrosis (CSIPF): A New Phenotype or a Fortuitous Overlap? Scoping Review and Case Series. J. Clin. Med. 2022, 11, 2065. [Google Scholar] [CrossRef]
- Pereira, J.O.; Fernandes, V.; Alfaro, T.M.; Freitas, S.; Cordeiro, C.R. Diagnosis of Fibrotic Hypersensitivity Pneumonitis: Is There a Role for Biomarkers? Life 2023, 13, 565. [Google Scholar] [CrossRef]
- Rafique, M.; Arslan, F.; Khan, J.; Zaki, S.; Hussain, A. Hypersensitivity Pneumonitis: An Interesting Case of Acute Shortness of Breath in a Young Patient. Cureus 2024, 16, e68683. [Google Scholar] [CrossRef]
- Rea, G.; Bocchino, M.; Lieto, R.; Ledda, R.E.; D’Alto, M.; Sperandeo, M.; Lucci, R.; Pasquinelli, P.; Sanduzzi Zamparelli, S.; Bocchini, G.; et al. The Unveiled Triad: Clinical, Radiological and Pathological Insights into Hypersensitivity Pneumonitis. J. Clin. Med. 2024, 13, 797. [Google Scholar] [CrossRef]
- Rittig, A.H.; Hilberg, O.; Ibsen, R.; Løkke, A. Incidence, Comorbidity and Survival Rate of Hypersensitivity Pneumonitis: A National Population-Based Study. ERJ Open Res. 2019, 5, 00259–02018. [Google Scholar] [CrossRef]
- Alberti, M.L.; Rincon-Alvarez, E.; Buendia-Roldan, I.; Selman, M. Hypersensitivity Pneumonitis: Diagnostic and Therapeutic Challenges. Front. Med. 2021, 8, 718299. [Google Scholar] [CrossRef]
- Cîrjaliu, R.E.; Gurrala, S.V.; Nallapati, B.; Krishna, V.; Oancea, C.; Tudorache, E.; Marc, M.; Bratosin, F.; Bogdan, I.; Rosca, O.; et al. Prognostic Implications of Initial Radiological Findings of Pulmonary Fibrosis in Patients with Acute SARS-CoV-2 Infection: A Prospective Multicentric Study. Diseases 2024, 12, 285. [Google Scholar] [CrossRef]
- Guler, S.A.; Wohlfarth, E.; Berezowska, S.; Geiser, T.K.; Ebner, L.; Funke-Chambour, M. Performance of a diagnostic algorithm for fibrotic hypersensitivity pneumonitis. A case-control study. Respir. Res. 2021, 22, 120. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Travis, W.D.; Lynch, D.A.; Brown, K.K.; Johannson, K.A.; Selman, M.; Ryu, J.H.; Wells, A.U.; Tony Huang, Y.C.; Pereira, C.A.C.; et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest 2021, 160, e97–e156. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef]
- Hamblin, M.; Prosch, H.; Vašáková, M. Diagnosis, course and management of hypersensitivity pneumonitis. Eur. Respir. Rev. 2022, 31, 210169. [Google Scholar] [CrossRef]
- Akkale, T.; Sarı, G.; Şimşek, C. Occupational hypersensitivity pneumonia. Tuberk. Toraks 2023, 71, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Trushenko, N.V.; Suvorova, O.A.; Pershina, E.S.; Nekludova, G.V.; Chikina, S.Y.; Levina, I.A.; Chernyaev, A.L.; Samsonova, M.V.; Tyurin, I.E.; Mustafina, M.K.; et al. Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases. Life 2023, 13, 467. [Google Scholar] [CrossRef]
- Amati, F.; Stainer, A.; Polelli, V.; Mantero, M.; Gramegna, A.; Blasi, F.; Aliberti, S. Efficacy of Pirfenidone and Nintedanib in Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 7849. [Google Scholar] [CrossRef] [PubMed]
- Ntiamoah, P.; Mehta, A.C. Beyond the Graft: Recurrence of Interstitial Lung Diseases Post Transplant. J. Clin. Med. 2025, 14, 1093. [Google Scholar] [CrossRef] [PubMed]
- Nosotti, M.; Leiva-Juarez, M.; D’Ovidio, F.; Van Raemdonck, D.; Ceulemans, L.; Keshavjee, S.; Rackauskas, M.; Paladini, P.; Luzzi, L.; Casado, P.M.; et al. Survival after lung transplantation for chronic hypersensitivity pneumonitis: Results from a large international cohort study. Transpl. Int. 2022, 35, 10450. [Google Scholar] [CrossRef]
- Cano-Jiménez, E.; Villar Gómez, A.; Velez Segovia, E.; Aburto Barrenechea, M.; Sellarés Torres, J.; Francesqui, J.; Portillo Carroz, K.; Solis Solis, A.J.; Acosta Fernández, O.; Llanos González, A.B.; et al. Prognostic factors of progressive fibrotic hypersensitivity pneumonitis: A large, retrospective, multicentre, observational cohort study. ERJ Open Res. 2024, 10, 00405–02023. [Google Scholar] [CrossRef]
- Ahmad, Y.; Mooney, J.; Allen, I.E.; Seaman, J.; Kalra, A.; Muelly, M.; Reicher, J. A Machine Learning System to Indicate Diagnosis of Idiopathic Pulmonary Fibrosis Non-Invasively in Challenging Cases. Diagnostics 2024, 14, 830. [Google Scholar] [CrossRef]
- Ruaro, B.; Pozzan, R.; Confalonieri, P.; Tavano, S.; Hughes, M.; Matucci Cerinic, M.; Baratella, E.; Zanatta, E.; Lerda, S.; Geri, P.; et al. Gastroesophageal Reflux Disease in Idiopathic Pulmonary Fibrosis: Viewer or Actor? To Treat or Not to Treat? Pharmaceuticals 2022, 15, 1033. [Google Scholar] [CrossRef]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Harhay, M.O.; Hayes, D.; Hsich, E.; Meiser, B.; Potena, L.; Robinson, A.; Rossano, J.W.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult heart transplantation report—2019; Focus theme: Donor and recipient size match. J. Heart Lung Transpl. 2019, 38, 1056–1066. [Google Scholar] [CrossRef]
- Churg, A.; Tazelaar, H.; Matej, R.; Vasakova, M.K.; Stewart, B.; Patel, D.; Duarte, E.; Gomez Manjarres, D.C.; Mehta, H.J.; Wright, J.L. Pathologic criteria for the diagnosis of usual interstitial pneumonia vs fibrotic hypersensitivity pneumonitis in transbronchial cryobiopsies. Mod. Pathol. 2023, 36, 100221. [Google Scholar] [CrossRef]
- Tinoco, E.M.; Bermudo, G.; Vicens-Zygmunt, V.; Luburich, P.; Llatjós, R.; Molina-Molina, M. Hypersensitivity Pneumonitis, a Differential Diagnosis of Cystic Lung Diseases. Pulmonology 2023, 29, 347–349. [Google Scholar] [CrossRef]
- Vindis, K.; Nemeth, N.; Marge, C.; Pantis, C.; Pop, M.G.; Pop, M.S.; Bondar, L.I.; Jurcau, M.C.; Babeș, K. Effects of Physical Exercise on Walking Distance and Functional Limitations in Patients with Chronic Dyspnea. Medicina 2025, 61, 636. [Google Scholar] [CrossRef]
- Leone, P.M.; Richeldi, L. Current Diagnosis and Management of Hypersensitivity Pneumonitis. Tuberc. Respir. Dis. 2020, 83, 122–131. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Ageely, G.; Souza, C.; De Boer, K.; Zahra, S.; Gomes, M.; Voduc, N. The Impact of Multidisciplinary Discussion (MDD) in the Diagnosis and Management of Fibrotic Interstitial Lung Diseases. Can. Respir. J. 2020, 2020, 9026171. [Google Scholar] [CrossRef]
- Gredic, M.; Karnati, S.; Ruppert, C.; Guenther, A.; Avdeev, S.N.; Kosanovic, D. Combined Pulmonary Fibrosis and Emphysema: When Scylla and Charybdis Ally. Cells 2023, 12, 1278. [Google Scholar] [CrossRef]
- Intra, J.; Biffi, A.; Basta, F.; Delfini, C.; Novati, N.; Zucchetti, E.; Luppi, F.; Casati, M. The Role of Serum IgG Precipitins against Six Typical Organic Antigens Involved in Hypersensitivity Pneumonitis: A 10-Year Retrospective Study of a Referral Interstitial Lung Disease Centre. Int. J. Transl. Med. 2024, 4, 381–386. [Google Scholar] [CrossRef]
- Shalmon, T.; Freund, O.; Wand, O.; Schneer, S.; Hershko, T.; Hadad, Y.; Aviram, G.; Bar-Shai, A.; Adir, Y.; Shitrit, D.; et al. Hypersensitivity pneumonitis radiologic features in interstitial lung diseases. Respir. Med. 2025, 236, 107901. [Google Scholar] [CrossRef]
- Moua, T.; Petnak, T.; Charokopos, A.; Baqir, M.; Ryu, J.H. Challenges in the Diagnosis and Management of Fibrotic Hypersensitivity Pneumonitis: A Practical Review of Current Approaches. J. Clin. Med. 2022, 11, 1473. [Google Scholar] [CrossRef]
- Sobiecka, M.; Szturmowicz, M.; Lewandowska, K.B.; Barańska, I.; Zimna, K.; Łyżwa, E.; Dybowska, M.; Langfort, R.; Radwan-Röhrenschef, P.; Roży, A.; et al. Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis. Diagnostics 2023, 13, 935. [Google Scholar] [CrossRef]
- Barkas, G.I.; Daniil, Z.; Kotsiou, O.S. The Role of Small Airway Disease in Pulmonary Fibrotic Diseases. J. Pers. Med. 2023, 13, 1600. [Google Scholar] [CrossRef] [PubMed]
- Vongvivitpatana, T.S.; Nambiar, A.M. A low forced vital capacity (FVC)/diffusing capacity of the lung for carbon monoxide (DLCO) ratio increases clinical suspicion for fibrotic hypersensitivity pneumonitis (FHP) over idiopathic pulmonary fibrosis (IPF). Cureus 2024, 16, e73008. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, K.; Lewandowska, K.B.; Kowalik, A.; Bartoszuk, I.; Radwan-Röhrenschef, P.; Sobiecka, M.; Dybowska, M.; Tomkowski, W.Z.; Szturmowicz, M. Does a Type of Inciting Antigen Correlate with the Presence of Lung Fibrosis in Patients with Hypersensitivity Pneumonitis? J. Clin. Med. 2024, 13, 5074. [Google Scholar] [CrossRef]
- Lassandro, G.; Picchi, S.G.; Corvino, A.; Massimo, C.; Tamburrini, S.; Vanore, L.; Urraro, G.; Russo, G.; Lassandro, F. Noninfectious Granulomatous Lung Disease: Radiological Findings and Differential Diagnosis. J. Pers. Med. 2024, 14, 134. [Google Scholar] [CrossRef]
- Feng, N.; Yasukawa, L.L.; Sen, A.; Greenberg, H.B. Permissive replication of homologous murine rotavirus in the mouse intestine is primarily regulated by VP4 and NSP1. J. Virol. 2013, 87, 8307–8316. [Google Scholar] [CrossRef]
- Bhatta, R.; Abou-Ghaida, J.; Bhattarai, S.; Blavo, C. A Case of Immunomodulator-Responsive Hypersensitivity Pneumonitis Secondary to Chronic Passive Smoke Inhalation. Cureus 2024, 16, e58723. [Google Scholar] [CrossRef]
- Sumi, T.; Takahashi, T.; Michimata, H.; Nagayama, D.; Koshino, Y.; Watanabe, H.; Yamada, Y.; Kodama, K.; Nishikiori, H.; Chiba, H. Exacerbation of Hypersensitivity Pneumonitis Induced by COVID-19. QJM 2023, 116, 235–236. [Google Scholar] [CrossRef]
- Fukihara, J.; Kondoh, Y. COVID-19 and interstitial lung diseases: A multifaceted look at the relationship between the two diseases. Respir. Investig. 2023, 61, 601–617. [Google Scholar] [CrossRef]
- Leong, S.W.; Bos, S.; Lordan, J.L.; Nair, A.; Fisher, A.J.; Meachery, G. Lung Transplantation for Interstitial Lung Disease: Evolution over Three Decades. BMJ Open Respir. Res. 2023, 10, e001387. [Google Scholar] [CrossRef]
- Irina, B.P.; Steluta, M.M.; Emanuela, T.; Diana, M.; Cristina, O.D.; Mirela, F.; Cristian, O. Respiratory Muscle Training Program Supplemented by a Cell-Phone Application in COPD Patients with Severe Airflow Limitation. Respir. Med. 2021, 190, 106679. [Google Scholar] [CrossRef]
- Cameli, P.; Alonzi, V.; d’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini, P.; Bargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Kypreos, M.; Barbera, T.; Newton, C.A.; Glazer, C.S.; Adams, T.N. Addition of antifibrotic therapy to immunosuppression in hypersensitivity pneumonitis: A case series. Respir. Med. Case Rep. 2021, 34, 101562. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mumtaz, S.; Baig, H.Z.; Mira-Avendano, I.; Wang, B.; Rojas, C.A.; Stowell, J.T.; Lesser, E.R.; Borkar, S.R.; Majithia, V.; et al. Longitudinal Study of Patients with Connective Tissue Disease–Interstitial Lung Disease and Response to Mycophenolate Mofetil and Rituximab. Diagnostics 2024, 14, 2702. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Tzouvelekis, A.; Bouros, E.; Karampitsakos, T.; Ntassiou, M.; Avdoula, E.; Trachalaki, A.; Antoniou, K.; Raghu, G.; Bouros, D. Clinical experience with antifibrotics in fibrotic hypersensitivity pneumonitis: A 3-year real-life observational study. ERJ Open Res. 2020, 6, 00152–02020. [Google Scholar] [CrossRef]
- Lewandowska, K.B.; Barańska, I.; Sobiecka, M.; Radwan-Rohrenschef, P.; Dybowska, M.; Franczuk, M.; Roży, A.; Skoczylas, A.; Bestry, I.; Kuś, J.; et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis. Diagnostics 2022, 12, 2767. [Google Scholar] [CrossRef]
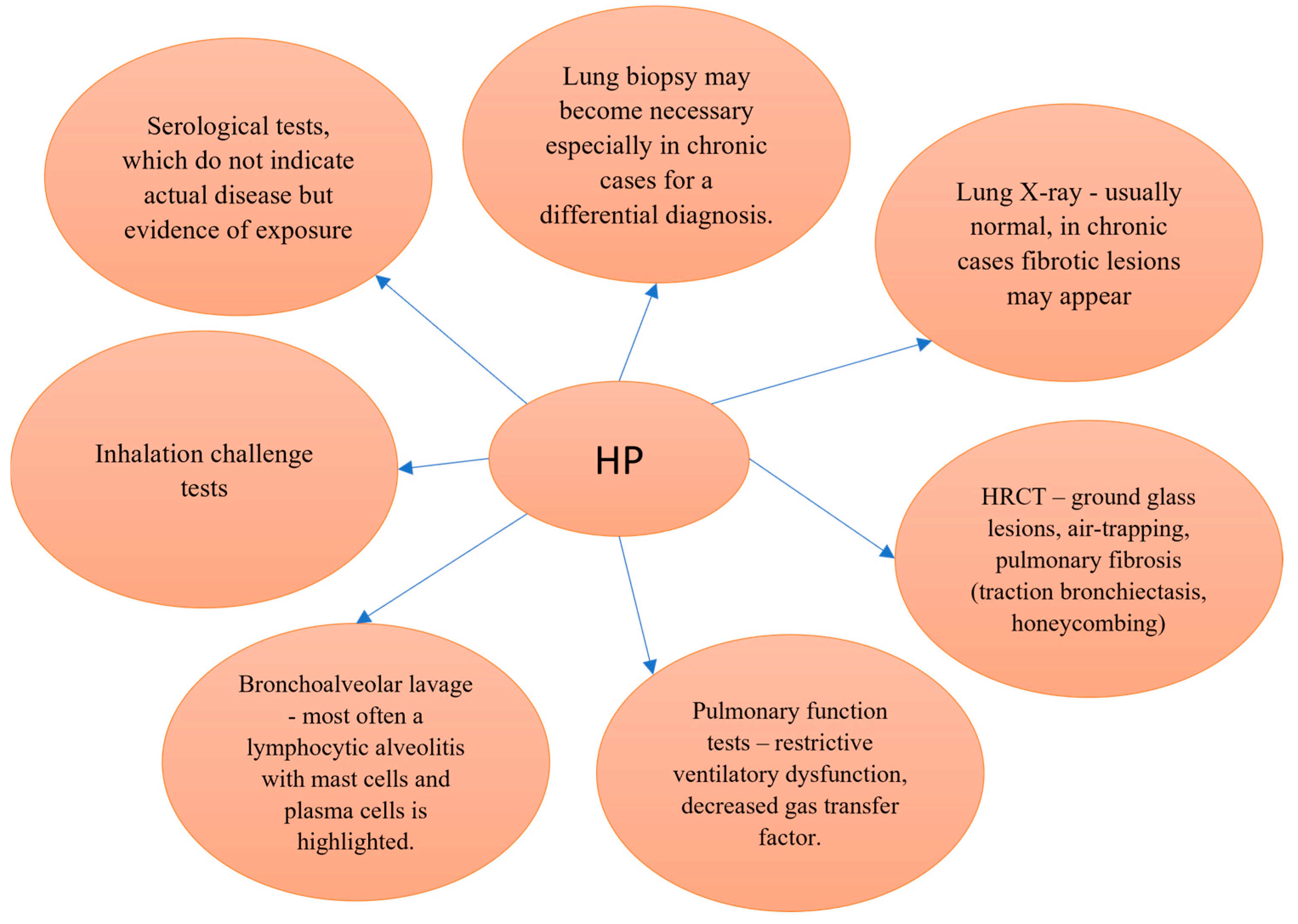
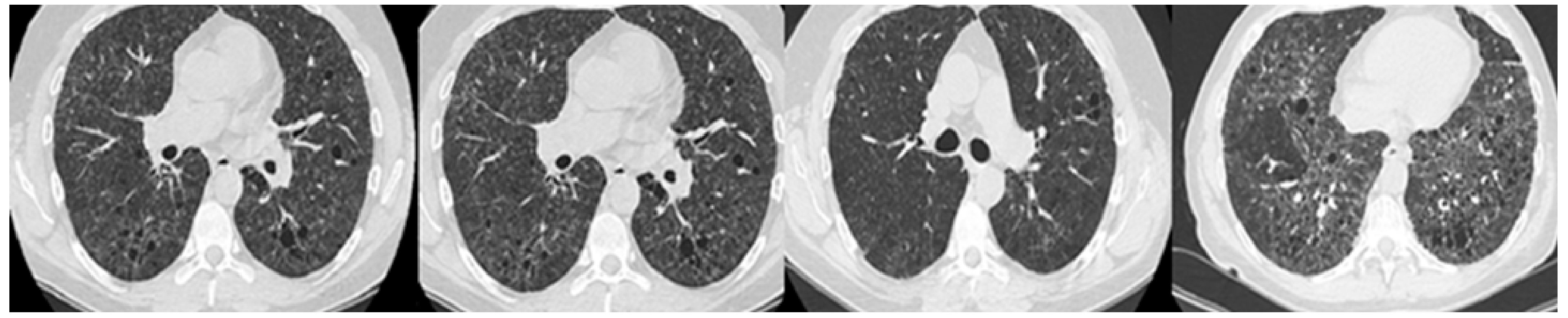
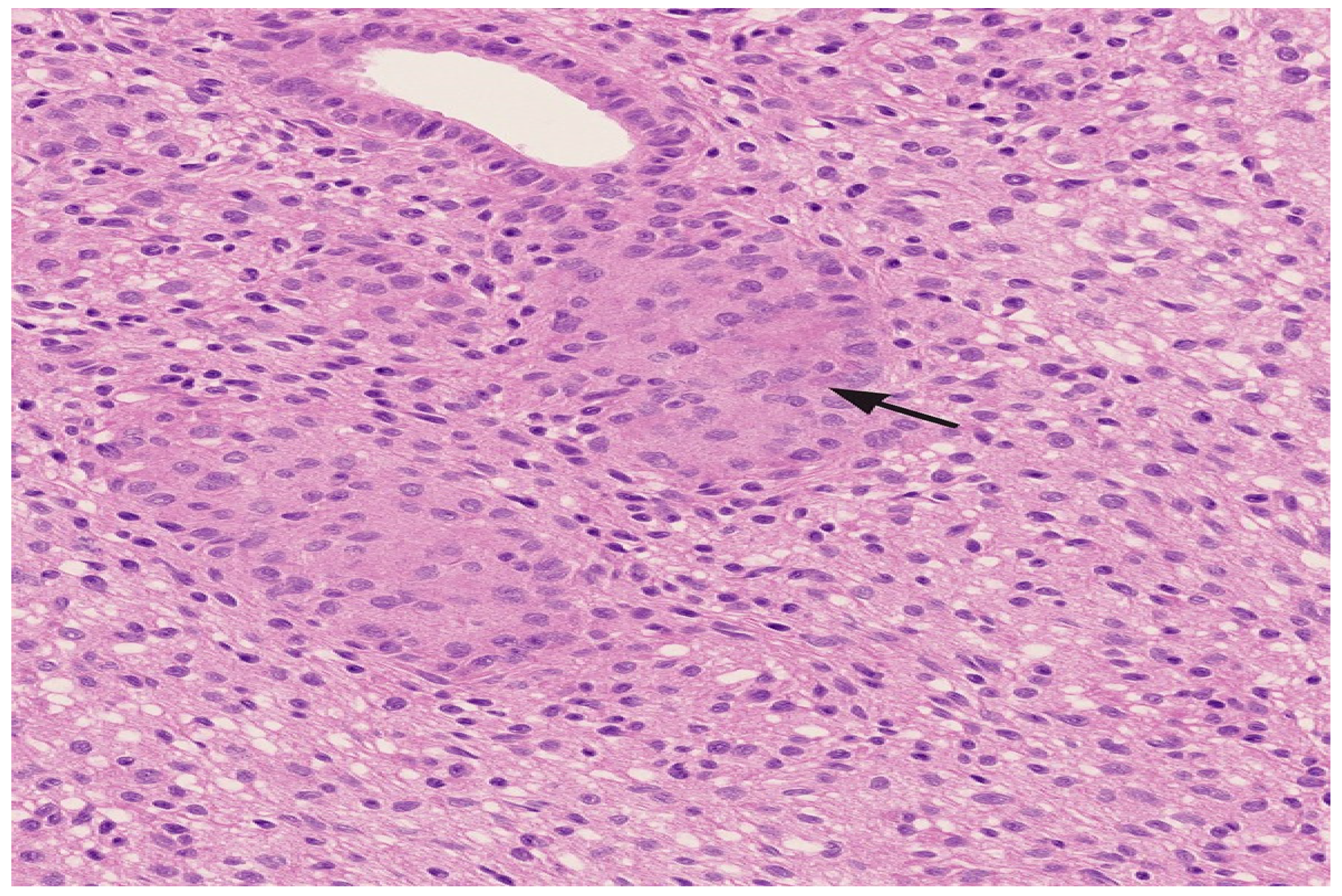
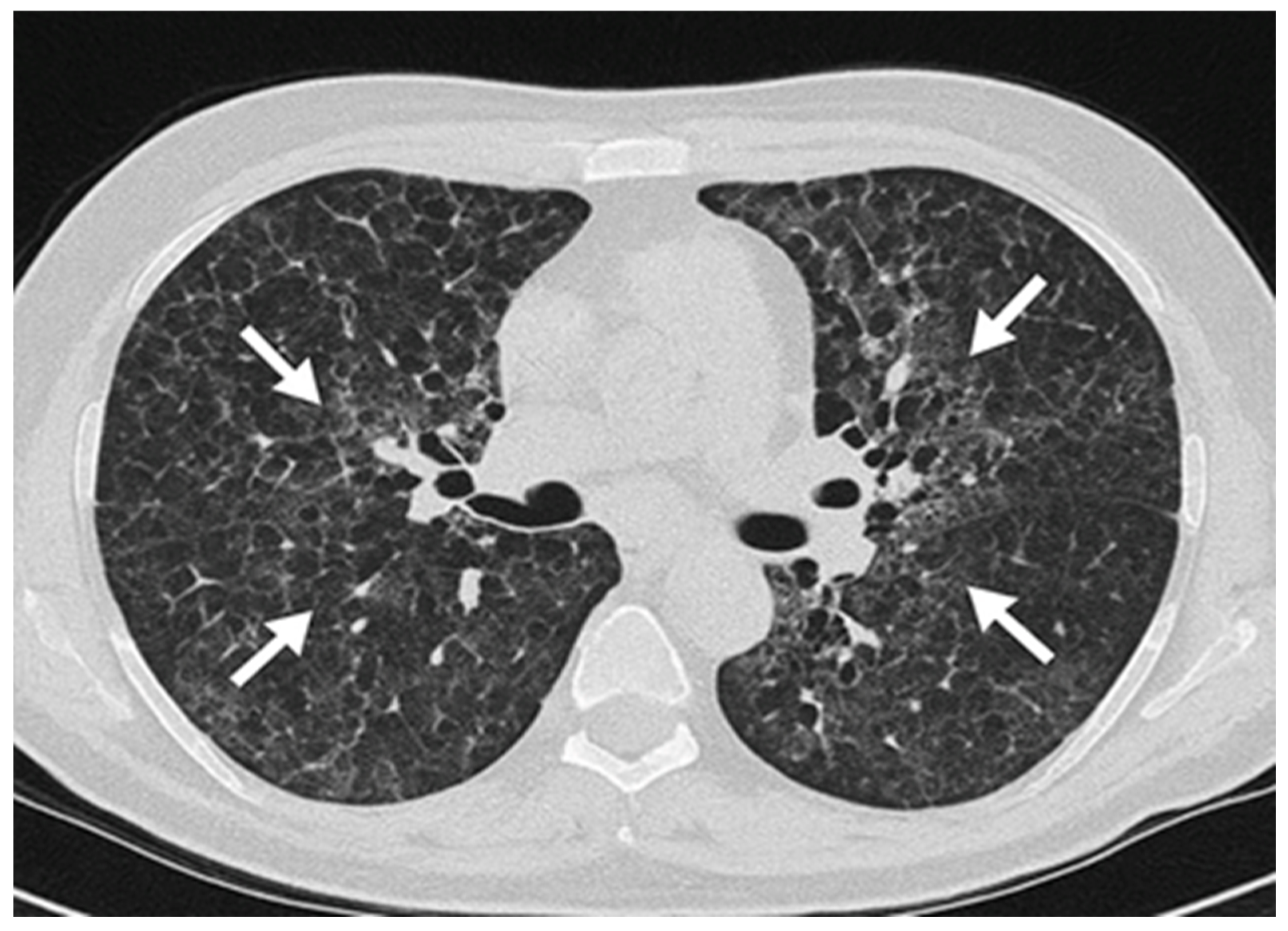

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diffusion Capacity and Lung Volumes/Pulmonary Function Test | ||||||
|---|---|---|---|---|---|---|
| Data | FVC % | DLCO mmol/(min*kPa) | TLC L | RV/TLC % | ERV % | RV % |
| 2018 | 80 | 42 | 85 | 70 | 185 | 58 |
| 2020 | 78 | 38 | 75 | 50 | 179 | 55 |
| 2022 | 109 | 36 | 74 | 38 | 191 | 50 |
| 2024 | 93 | 32 | 75 | 62 | 177 | 49 |
| Antibody | Result | Reference Value |
|---|---|---|
| Anti-nRNP/Sm | Negative | (Negative) |
| Anti-Sm | Negative | (Negative) |
| Anti-SS-A (Ro) | Negative | (Negative) |
| Anti-Ro-52 | Negative | (Negative) |
| Anti-SS-B (La) | Negative | (Negative) |
| Anti-Scl-70 | Negative | (Negative) |
| Anti-PM-Scl 100 | Negative | (Negative) |
| Anti-Jo-1 | Negative | (Negative) |
| Anti-Centromere B | Negative | (Negative) |
| Anti-PCNA | Negative | (Negative) |
| Anti-dsDNA | Negative | (Negative) |
| Anti-Nucleosome | Negative | (Negative) |
| Anti-Histone | Negative | (Negative) |
| Anti-Ribosomal P Protein | Negative | (Negative) |
| Anti-AMA-M2 | Negative | (Negative) |
| Anti-DFS70 | Negative | (Negative) |
| CAP-Class Reference Ranges | ||
|---|---|---|
| CAP-Class | AU/mL Range | Interpretation |
| Class 0 | 0.10–0.35 | Negative |
| Class 1 | 0.35–0.70 | Threshold value |
| Class 2 | 0.70–3.50 | Weakly positive |
| Class 3 | 3.50–17.5 | Positive |
| Class 4 | 17.5–50.0 | Highly positive |
| Class 5 | 50.0–100 | Intensely positive |
| Class 6 | >100 | Exceptionally high value |
| Allergen-Specific IgE Results with CAP-Class Interpretation | ||
| Allergen | Result (AU/mL) | CAP-Class |
| Chicken feathers | <0.10 (Negative) | Class 0 |
| Chicken droppings | <0.10 (Negative) | Class 0 |
| Parameter | Value (On Room Air) |
|---|---|
| pH | 7.43 |
| PaO2 | 61 mmHg |
| PaCO2 | 36 mmHg |
| HCO3− | 23 mEq/L |
| SaO2 | 90% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mot, M.-D.; Olar, D.C.; Vulciu, P.A.; Barata, P.-I.; Bouros-Tataru, A.-L.; Butari, D.B.; Șandor, F.M.; Bondar, L.I. Fibrotic Hypersensitivity Pneumonitis: A Diagnostic Challenge Leading to Lung Transplantation. Diagnostics 2025, 15, 1267. https://doi.org/10.3390/diagnostics15101267
Mot M-D, Olar DC, Vulciu PA, Barata P-I, Bouros-Tataru A-L, Butari DB, Șandor FM, Bondar LI. Fibrotic Hypersensitivity Pneumonitis: A Diagnostic Challenge Leading to Lung Transplantation. Diagnostics. 2025; 15(10):1267. https://doi.org/10.3390/diagnostics15101267
Chicago/Turabian StyleMot, Maria-Daniela, Dana Cristina Olar, Paula Alexandra Vulciu, Paula-Irina Barata, Ana-Liana Bouros-Tataru, Denis Bogdan Butari, Florin Mihai Șandor, and Laura Ioana Bondar. 2025. "Fibrotic Hypersensitivity Pneumonitis: A Diagnostic Challenge Leading to Lung Transplantation" Diagnostics 15, no. 10: 1267. https://doi.org/10.3390/diagnostics15101267
APA StyleMot, M.-D., Olar, D. C., Vulciu, P. A., Barata, P.-I., Bouros-Tataru, A.-L., Butari, D. B., Șandor, F. M., & Bondar, L. I. (2025). Fibrotic Hypersensitivity Pneumonitis: A Diagnostic Challenge Leading to Lung Transplantation. Diagnostics, 15(10), 1267. https://doi.org/10.3390/diagnostics15101267