
Cardiac Involvement in Classical Organic Acidurias: Clinical Profile and Outcome in a Pediatric Cohort

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Definition of Cardiac Involvement
- -
- Dilated cardiomyopathy (DCM) was diagnosed in the presence of increased ventricular end-diastolic diameter (>2 Z-Score) associated with any degree of systolic dysfunction, defined as impaired left ventricular ejection fraction (EF). EF was measured by the biplane Simpson method (normal value: >55%, mild dysfunction: 45–54%, moderate dysfunction: 30–44%, severe dysfunction: <30%).
- -
- Evidence of supraventricular or ventricular arrhythmias.
- -
- Otherwise unexplained Qtc prolongation (>460 ms).
2.2. Statistical Analysis
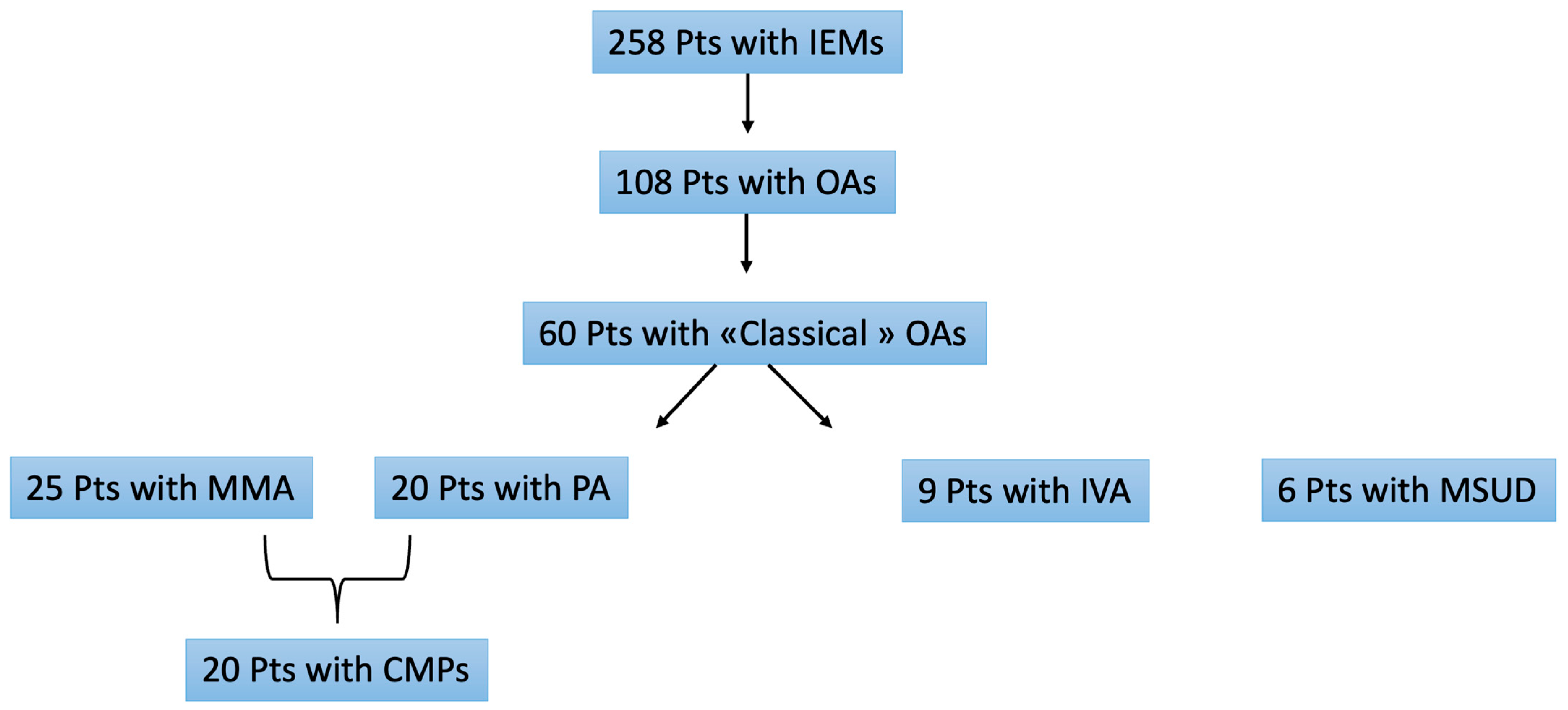
3. Results
3.1. Baseline Characteristics
3.2. Genetic Results
3.3. Extra-Cardiac Involvement
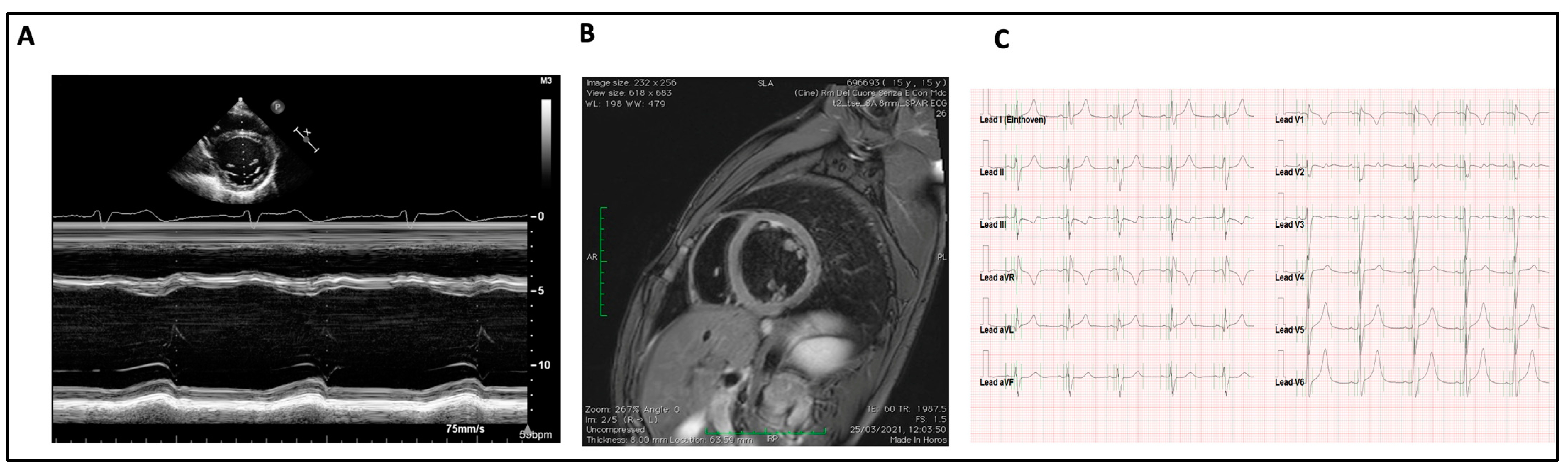
3.4. Cardiac Involvement
3.5. Management
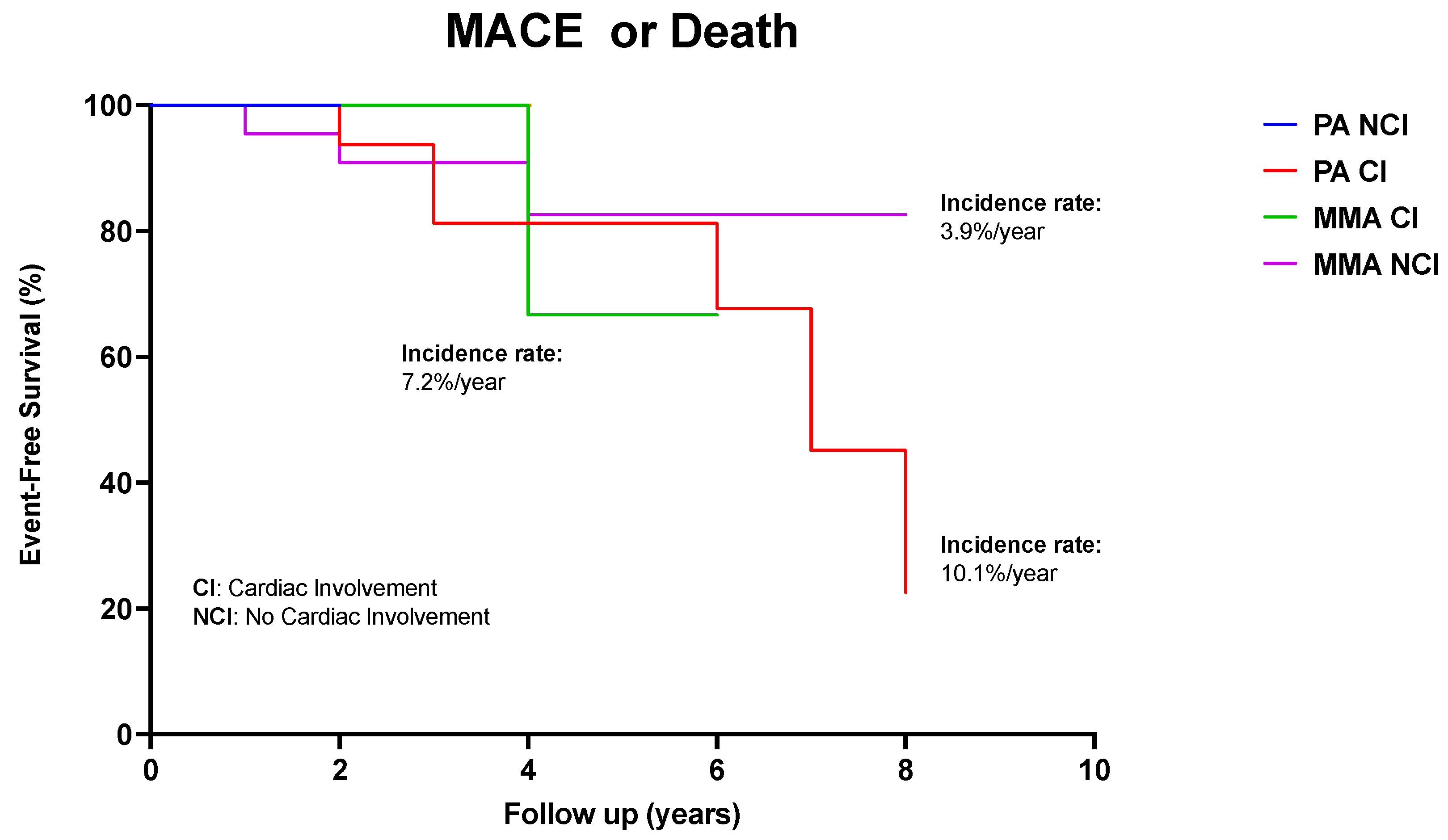
3.6. Long-Term Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scriver, R.; Beaudet, A.; Sly, E.S.; Valle, D. The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Waber, L. Inborn errors of metabolism. Pediatr. Ann. 1990, 19, 105–109, 112–113, 117–118. [Google Scholar] [CrossRef]
- Villani, G.R.; Gallo, G.; Scolamiero, E.; Salvatore, F.; Ruoppolo, M. “Classical organic acidurias”: Diagnosis and pathogenesis. Clin. Exp. Med. 2016, 17, 305–323. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Narayanan, M.P.; Vasudevan, D.M. Organic acidurias: An updated review. Indian J. Clin. Biochem. 2011, 26, 319–325. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in children: Classification and diagnosis: A scientific statement from the American heart association. Circulation 2019, 140, E9–E68. [Google Scholar] [CrossRef]
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef]
- Moss, A.J.; Kass, R.S. Long QT syndrome: From channels to cardiac arrhythmias. J. Clin. Investig. 2005, 115, 2018–2024. [Google Scholar] [CrossRef]
- Park, K.C.; Krywawych, S.; Richard, E.; Desviat, L.R.; Swietach, P. Cardiac Complications of Propionic and Other Inherited Organic Acidemias. Front. Cardiovasc. Med. 2020, 7, 617451. [Google Scholar] [CrossRef]
- Berry, G.T.; Blume, E.D.; Wessel, A.; Singh, T.; Hecht, L.; Marsden, D.; Sahai, I.; Elisofon, S.; Ferguson, M.; Kim, H.B.; et al. The reoccurrence of cardiomyopathy in propionic acidemia after liver transplantation. JIMD Rep. 2020, 54, 3–8. [Google Scholar] [CrossRef]
- Deodato, F.; Boenzi, S.; Santorelli, F.M.; Dionisi-Vici, C. Methylmalonic and propionic aciduria. Am. J. Med. Genet. Part C Semin. Med. Genet. 2006, 142, 104–112. [Google Scholar] [CrossRef]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Barić, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. additional individual contributors of the E-IMD consortium. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef]
- Tajima, G.; Kagawa, R.; Sakura, F.; Nakamura-Utsunomiya, A.; Hara, K.; Yuasa, M.; Hasegawa, Y.; Sasai, H.; Okada, S. Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant. Int. J. Neonatal Screen. 2021, 7, 35. [Google Scholar] [CrossRef]
- Baumgartner, D.; Scholl-Bürgi, S.; Sass, J.O.; Sperl, W.; Schweigmann, U.; Stein, J.I.; Karall, D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionicacidemia. J. Pediatr. 2007, 150, 192–197.e1. [Google Scholar] [CrossRef]
- Peuhkurinen, K.J.; Takala, T.E.; Nuutinen, E.M.; Hassinen, I.E. Tricarboxylic acid cycle metabolites during ischemia in isolated perfused rat heart. Am. J. Physiol. 1983, 244, H281–H288. [Google Scholar] [CrossRef]
- Mardach, R.; Verity, M.A.; Cederbaum, S.D. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol. Genet. Metab. 2005, 85, 286–290. [Google Scholar] [CrossRef]
- Gibala, M.J.; Young, M.E.; Taegtmeyer, H. Anaplerosis of the citric acid cycle: Role in energy mtabolism of heart and skeletal muscle. Acta Physiol. Scand. 2000, 168, 657–665. [Google Scholar] [CrossRef]
- Szymańska, E.; Jezela-Stanek, A.; Bogdańska, A.; Rokicki, D.; Ehmke vel Emczyńska-Seliga, E.; Pajdowska, M.; Ciara, E.; Tylki-Szymańska, A. Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria. Diagnostics 2020, 10, 738. [Google Scholar] [CrossRef]
- Sun, B.; Leem, C.H.; Vaughan-Jones, R.D. Novel chloride-dependent acid loader in the guinea-pig ventricular myocyte: Part of a dual acid-loading mechanism. J. Physiol. 1996, 495, 65–82. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Romano, S.; Valayannopoulos, V.; Touati, G.; Jais, J.P.; Rabier, D.; de Keyzer, Y.; Bonnet, D.; de Lonlay, P. Cardiomyopathies in propionic aciduria arereversible after liver transplantation. J. Pediatr. 2010, 156, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Ameloot, K.; Vlasselaers, D.; Dupont, M.; Meersseman, W.; Desmet, L.; Vanhaecke, J.; Vermeer, N.; Meyns, B.; Pirenne, J.; Cassiman, D.; et al. Left ventricular assist device as bridge to liver transplantation in a patient with propionic acidemia and cardiogenic shock. J. Pediatr. 2011, 158, 866–867. [Google Scholar] [CrossRef]
- Vockley, J. Disorders of Branched Chain Amino and Organic Acid Metabolism. In Rudolph’s Pediatrics, 23rd ed.; Kline, M.W., Ed.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Manoli, I.; Venditti, C.P. Disorders of branched chain amino acid metabolism. Transl. Sci. Rare Dis. 2016, 1, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.M.; Touati, G.; Delonlay, P.; Jouvet, P.; Schlenzig, J.; Narcy, C.; Laurent, J.; Rabier, D.; Kamoun, P.; Jan, D.; et al. Liver transplantation in propionic acidaemia. Eur. J. Pediatr. 1999, 158, S065–S069. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Walter, J.H.; McKiernan, P.J. The management of organic acidaemias: The role of transplantation. J. Inherit. Metab. Dis. 2001, 24, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, A.; Garbade, S.; Hoffmann, G.; Gorenflo, M.; Kölker, S.; Staufner, C. Cardiac phenotype in propionic acidemia—Results of an observational monocentric study. Mol. Genet. Metab. 2020, 130, 41–48. [Google Scholar] [CrossRef]
- Shchelochkov, O.A.; Carrillo, N.; Venditti, C. Propionic acidemia. In Gene Reviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Yannicelli, S.; Acosta, P.B.; Velazquez, A.; Bock, H.-G.; Marriage, B.; Kurczynski, T.W.; Miller, M.; Korson, M.; Steiner, R.D.; Rutledge, L.; et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol. Genet. Metab. 2003, 80, 181–188. [Google Scholar] [CrossRef]
- Chandler, R.J.; Venditti, C.P. Gene therapy for organic acidemias: Lessons learned from methylmalonic and propionic acidemia. J. Inherit. Metab. Dis. 2023. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Family History |
|
| Clinical examination |
|
| Cardiologic evaluation |
|
| Laboratory investigation |
|
| Overall | PA | MMA | IVA | MSUD | |
|---|---|---|---|---|---|
| Number of patients | 60 | 20 (33.3%) | 25 (41.6%) | 9 (15%) | 6 (10%) |
| Male patients | 32 (53%) | 8 (40%) | 14 (56%) | 8 (88.9%) | 2 (33.4%) |
| Mean age of diagnosis of OAs | <1 | <1 | <1 | <1 | <1 |
| Family history | 7 (11.7%) | 4 (20%) | 1 (4%) | 2(22.2%) | 0 |
| NBS diagnosis | 17 (28.3%) | 2 (10%) | 8 (32%) | 4 (44.5%) | 3 (50%) |
| Clinical diagnosis | 36 (60%) | 14 (70%) | 16 (64%) | 3 (33.4%) | 3 (50%) |
| Molecular analysis | 42(70) | 12(60%) | 23 (92%) | 7(77.8%) | 0 |
| Neurological involvement | 28 (46.7%) | 12 (60%) | 8 (32%) | 5 (55.6%) | 3 (50%) |
| Ocular involvement | 5 (8.3%) | 2 (10%) | 2 (8%) | 0 | 1 (16.7%) |
| Renal involvement | 5 (8.3%) | 1 (5%) | 3 (12%) | 0 | 1 (16.7%) |
| Failure to thrive | 4 (6.7%) | 2 (10%) | 1 (4%) | 1 (11.2%) | 0 |
| Hematological involvement | 5 (8.3%) | 2 (10%) | 3 (12%) | 0 | 0 |
| Gastrointestinal | 7 (11.7%) | 2 (10%) | 3 (12%) | 1 (11.2%) | 1 (16.7%) |
| Skeletal involvement | 6 (10%) | 3 (15%) | 1 (4%) | 0 | 2 (33.4%) |
| Cardiac involvement | 23 (38%) | 17 (85%) | 6(24%) | 0 | 0 |
| CMP in CI | 20 (33.3%) | 17(85%) | 3 (12%) | 0 | 0 |
| Mean age of CMP onset | 10.3 | 10.2 | 11 | 0 | 0 |
| Average EF% at first valuation | 53.3 | 52.76 | 56.7 | 0 | 0 |
| Average EF% at last valuation | 46.9 | 46.4 | 50 | 0 | 0 |
| Average QTc (Sec) | 465.5 | 465.3 | 466.7 | 0 | 0 |
| MACEs with CI (%/year) | 9% | 10% | 7.2% | 0 | 0 |
| Death for HF | 2 (3.3%) | 2 (10%) | 0 | 0 | 0 |
| Hospitalization for HF | 4 (6.6%) | 4 (20%) | 0 | 0 | 0 |
| Metabolic Crisis | 4(6.6%) | 3 (15%) | 1 (4%) | 0 | 0 |
| Liver transplantation | 7 (11.7%) | 5 (25%) | 2(8%) | 0 | 0 |
| Mortality | 5 (8,3%) | 4 (20%) | 1 (4%) | 0 | 0 |
| Cardiac cause | 2 (10%) | 2 (10%) | 0 | 0 | 0 |
| Non-cardiac cause | 3 (15%) | 2 (10%) | 1 (4%) | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passantino, S.; Chiellino, S.; Girolami, F.; Zampieri, M.; Calabri, G.B.; Spaziani, G.; Bennati, E.; Porcedda, G.; Procopio, E.; Olivotto, I.; et al. Cardiac Involvement in Classical Organic Acidurias: Clinical Profile and Outcome in a Pediatric Cohort. Diagnostics 2023, 13, 3674. https://doi.org/10.3390/diagnostics13243674
Passantino S, Chiellino S, Girolami F, Zampieri M, Calabri GB, Spaziani G, Bennati E, Porcedda G, Procopio E, Olivotto I, et al. Cardiac Involvement in Classical Organic Acidurias: Clinical Profile and Outcome in a Pediatric Cohort. Diagnostics. 2023; 13(24):3674. https://doi.org/10.3390/diagnostics13243674
Chicago/Turabian StylePassantino, Silvia, Serena Chiellino, Francesca Girolami, Mattia Zampieri, Giovanni Battista Calabri, Gaia Spaziani, Elena Bennati, Giulio Porcedda, Elena Procopio, Iacopo Olivotto, and et al. 2023. "Cardiac Involvement in Classical Organic Acidurias: Clinical Profile and Outcome in a Pediatric Cohort" Diagnostics 13, no. 24: 3674. https://doi.org/10.3390/diagnostics13243674
APA StylePassantino, S., Chiellino, S., Girolami, F., Zampieri, M., Calabri, G. B., Spaziani, G., Bennati, E., Porcedda, G., Procopio, E., Olivotto, I., & Favilli, S. (2023). Cardiac Involvement in Classical Organic Acidurias: Clinical Profile and Outcome in a Pediatric Cohort. Diagnostics, 13(24), 3674. https://doi.org/10.3390/diagnostics13243674