
Current Challenges in the Diagnosis of Pediatric Cutaneous Mastocytosis

 ,
,  and
and
Abstract
:1. Introduction
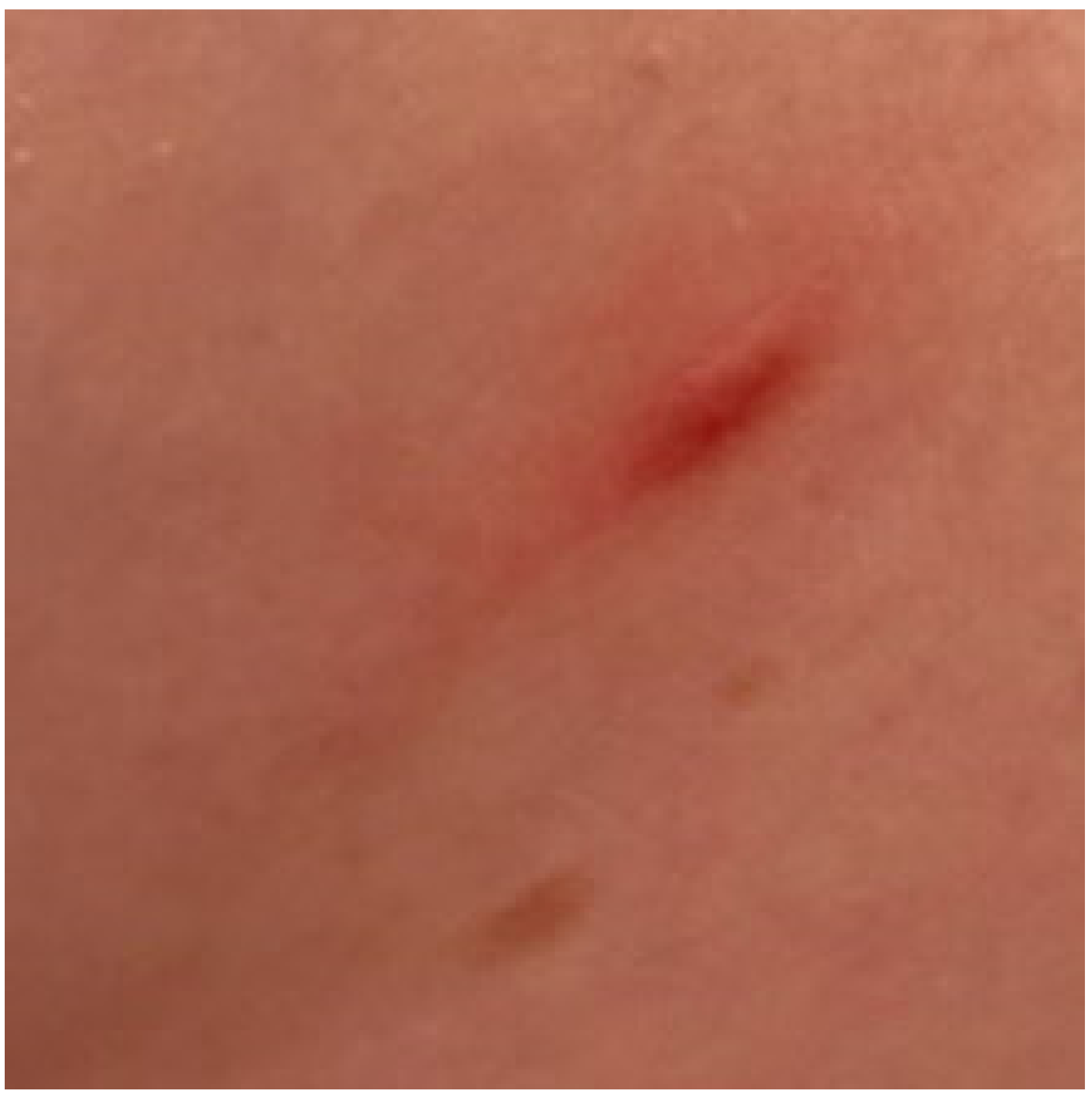
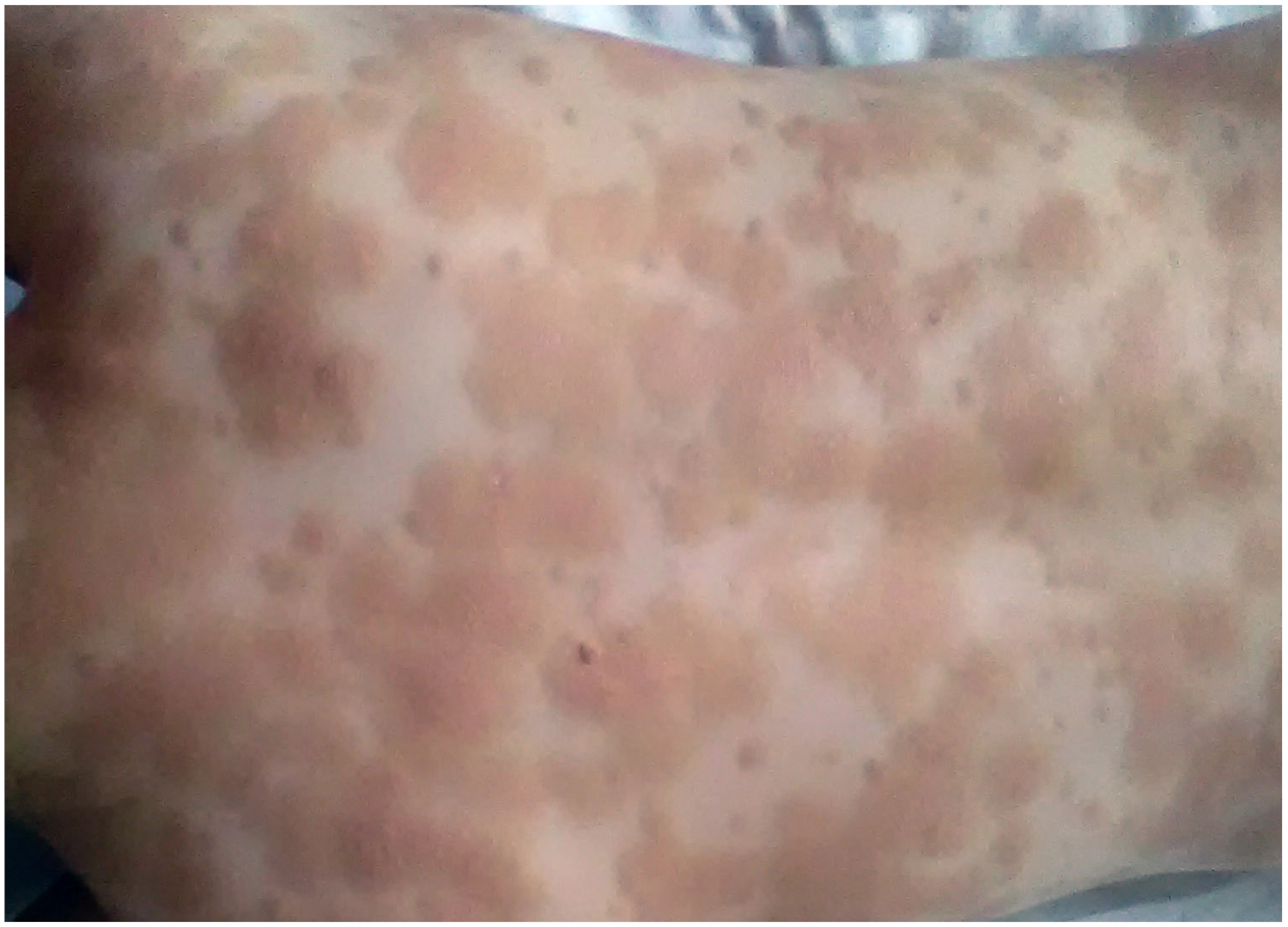
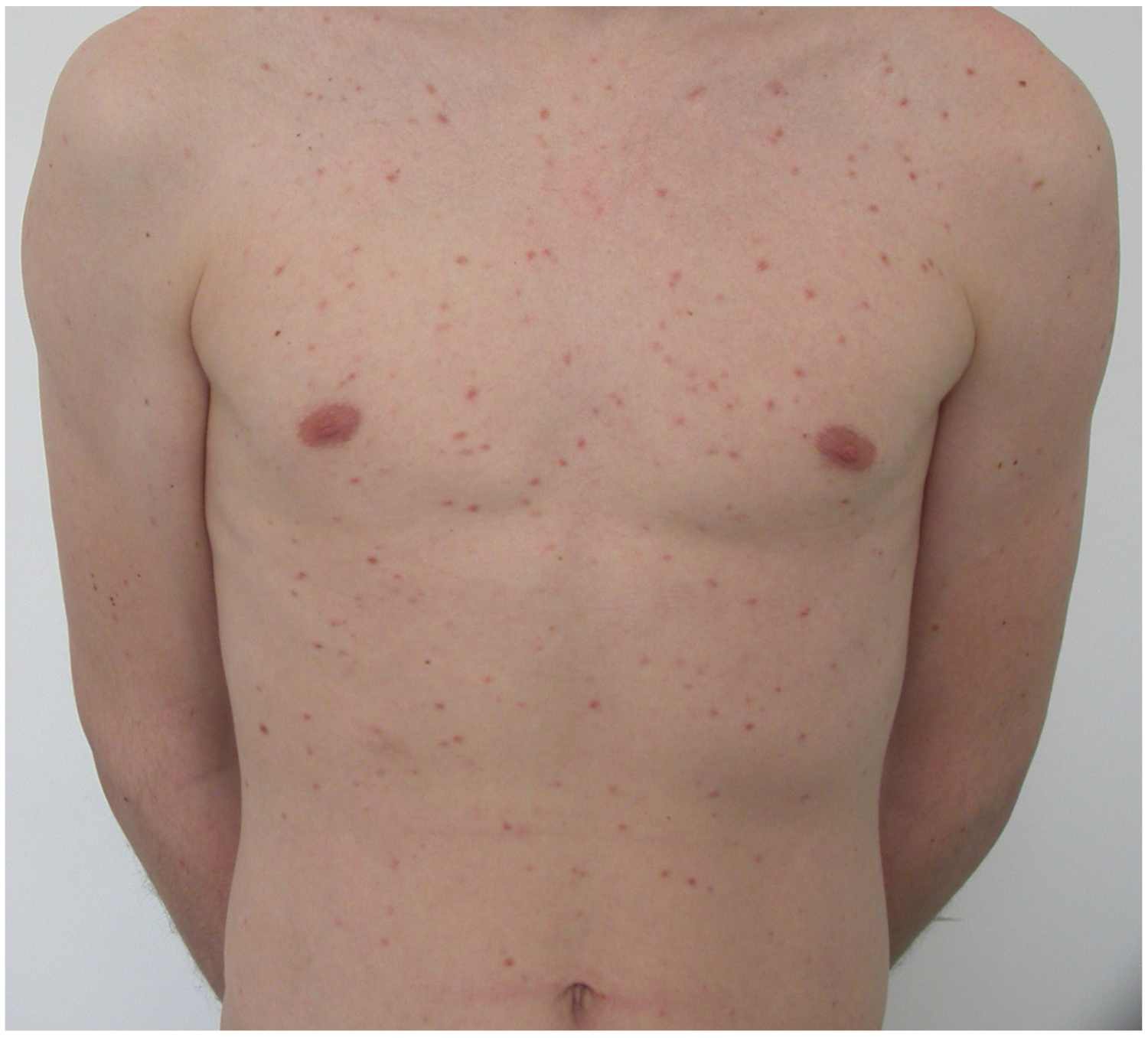
2. Clinical Characteristic of Skin Involvement in Children with CM
3. Mast Cell Mediator-Related Symptoms and Anaphylaxis in Children with CM
4. Diagnostic Evaluation in Children with Suspected Mastocytosis
4.1. Skin Histology
4.2. Molecular Testing for Activating KIT Mutations in the Skin and Peripheral Blood
4.3. Serum Tryptase Level
4.4. Screening for Hereditary Alpha Tryptasemia
5. Differential Diagnosis in Children with Suspected CM
5.1. Differential Diagnosis of MPCM
5.2. Differential Diagnosis of DCM
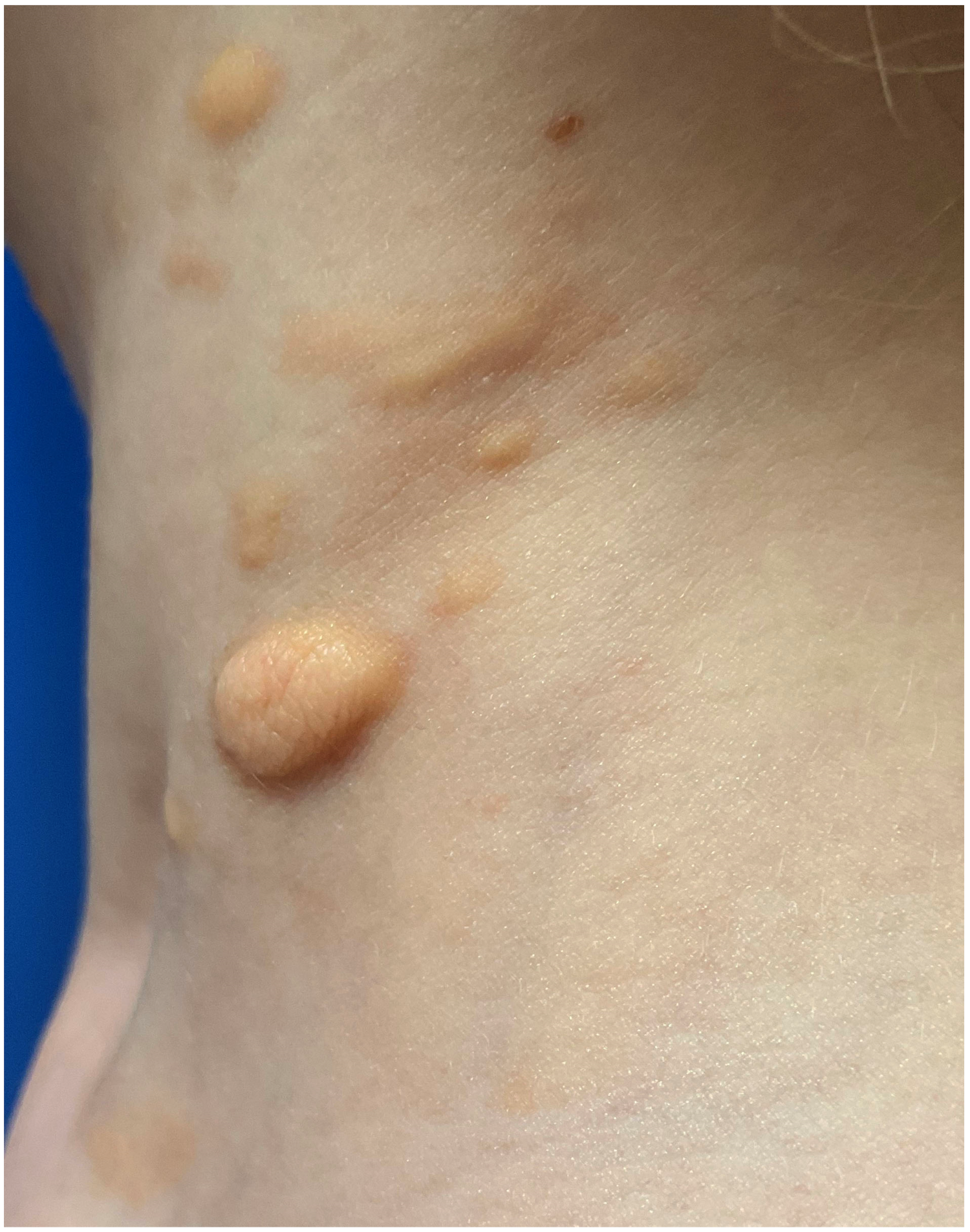
5.3. Differential Diagnosis of Mastocytoma
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Akin, C.; Sperr, W.R.; Horny, H.-P.; Arock, M.; Metcalfe, D.D.; Galli, S.J. New Insights into the Pathogenesis of Mastocytosis: Emerging Concepts in Diagnosis and Therapy. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 361–386. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. Systemic Mastocytosis in Adults: 2023 Update on Diagnosis, Risk Stratification and Management. Am. J. Hematol. 2023, 98, 1097–1116. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Hemasphere 2021, 5, e646. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 Updated WHO Classification and Novel Emerging Treatment Concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Cutaneous Manifestations in Patients with Mastocytosis: Consensus Report of the European Competence Network on Mastocytosis; The American Academy of Allergy, Asthma & Immunology; And the European Academy of Allergology and Clinical Immunology. J. Allergy Clin. Immunol. 2016, 137, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Hartmann, K.; Carter, M.C.; Siebenhaar, F.; Alvarez-twose, I.; Torrado, I.; Brockow, K.; Renke, J.; Irga-jaworska, N.; Plata-nazar, K.; et al. Molecular Background, Clinical Features and Management of Pediatric Mastocytosis: Status 2021. Int. J. Mol. Sci. 2021, 22, 2586. [Google Scholar] [CrossRef]
- Tiano, R.; Krase, I.Z.; Sacco, K. Updates in Diagnosis and Management of Paediatric Mastocytosis. Curr. Opin. Allergy Clin. Immunol. 2023, 23, 158–163. [Google Scholar] [CrossRef]
- Renke, J.; Irga-Jaworska, N.; Lange, M. Pediatric and Hereditary Mastocytosis. Immunol. Allergy Clin. N. Am. 2023, 43, 665–679. [Google Scholar] [CrossRef]
- Schaffer, J.V. Pediatric Mastocytosis: Recognition and Management. Am. J. Clin. Dermatol. 2021, 22, 205–220. [Google Scholar] [CrossRef]
- Polivka, L.; Rossignol, J.; Neuraz, A.; Condé, D.; Agopian, J.; Méni, C.; Garcelon, N.; Dubreuil, P.; Maouche-Chrétien, L.; Hadj-Rabia, S.; et al. Criteria for the Regression of Pediatric Mastocytosis: A Long-Term Follow-Up. J. Allergy Clin. Immunol. Pract. 2021, 9, 1695–1704.e5. [Google Scholar] [CrossRef] [PubMed]
- Méni, C.; Bruneau, J.; Georgin-Lavialle, S.; Le Saché De Peufeilhoux, L.; Damaj, G.; Hadj-Rabia, S.; Fraitag, S.; Dubreuil, P.; Hermine, O.; Bodemer, C. Paediatric Mastocytosis: A Systematic Review of 1747 Cases. Br. J. Dermatol. 2015, 172, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Gogia, A.; Sharawat, S.K.; Kumar, R.; Sarkar, C.; Bakhshi, S. Systemic Mastocytosis Associated With Childhood Acute Myeloid Leukemia. J. Pediatr. Hematol. Oncol. 2013, 35, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Nong, L.; Liang, L.; Wang, W.; Li, T. De Novo Mast Cell Leukemia without CD25 Expression and KIT Mutations: A Rare Case Report in a 13-Year-Old Child. Diagn. Pathol. 2018, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Monnier, J.; Georgin-Lavialle, S.; Canioni, D.; Lhermitte, L.; Soussan, M.; Arock, M.; Bruneau, J.; Dubreuil, P.; Bodemer, C.; Chandesris, M.-O.; et al. Mast Cell Sarcoma: New Cases and Literature Review. Oncotarget 2016, 7, 66299–66309. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Arock, M.; Lyons, J.; Bachelot, G.; Schwartz, L.; Reiter, A.; Jawhar, M.; Schwaab, J.; Lange, M.; Greiner, G.; et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 411. [Google Scholar] [CrossRef] [PubMed]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating C-KIT Mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef]
- Arase, N.; Wataya-Kaneda, M.; Murota, H.; Nakagawa, Y.; Yamaoka, T.; Itoi-Ochi, S.; Hirayasu, K.; Arase, H.; Fujimoto, M.; Katayama, I. Genotype and Phenotype Analysis of Patients with Pediatric Cutaneous Mastocytosis, Especially Wild-type KIT Patients. J. Dermatol. 2020, 47, 426–429. [Google Scholar] [CrossRef]
- Valent, P.; Spanblöchl, E.; Sperr, W.; Sillaber, C.; Zsebo, K.; Agis, H.; Strobl, H.; Geissler, K.; Bettelheim, P.; Lechner, K. Induction of Differentiation of Human Mast Cells from Bone Marrow and Peripheral Blood Mononuclear Cells by Recombinant Human Stem Cell Factor/Kit-Ligand in Long-Term Culture. Blood 1992, 80, 2237–2245. [Google Scholar] [CrossRef]
- Méni, C.; Georgin-Lavialle, S.; Le Saché de Peufeilhoux, L.; Jais, J.P.; Hadj-Rabia, S.; Bruneau, J.; Fraitag, S.; Hanssens, K.; Dubreuil, P.; Hermine, O.; et al. Paediatric Mastocytosis: Long-Term Follow-up of 53 Patients with Whole Sequencing of KIT. A Prospective Study. Br. J. Dermatol. 2018, 179, 925–932. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Liu, X.; Kang, L.; Liu, X. Genotypic and Phenotypic Characteristics of Chinese Neonates with Cutaneous Mastocytosis: A Case Report and Literature Review. J. Int. Med. Res. 2020, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wasąg, B.; Niedoszytko, M.; Piskorz, A.; Lange, M.; Renke, J.; Jassem, E.; Biernat, W.; Debiec-Rychter, M.; Limon, J. Novel, Activating KIT-N822I Mutation in Familial Cutaneous Mastocytosis. Exp. Hematol. 2011, 39, 859–865.e2. [Google Scholar] [CrossRef] [PubMed]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT Mutation Analysis in Mast Cell Neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.J.; Tharp, M.D. Comparison of Lesional Skin C-KIT Mutations with Clinical Phenotype in Patients with Mastocytosis. Clin. Exp. Dermatol. 2018, 43, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Shapiro, N.; Bhutada, A.; Rastogi, S. C-KIT-Positive Fatal Diffuse Cutaneous Mastocytosis with Systemic Manifestations in a Neonate. J. Pediatr. Hematol./Oncol. 2019, 41, e338–e340. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, H.A.; Lundgren, A.D.; Carter, M.C.; Diaz, L.Z.; Levy, M.L. Management of a Neonate with Diffuse Cutaneous Mastocytosis: Case Report and Literature Review. Pediatr. Dermatol. 2019, 36, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Morren, M.-A.; Hoppé, A.; Renard, M.; Debiec Rychter, M.; Uyttebroeck, A.; Dubreuil, P.; Martin, L. Imatinib Mesylate in the Treatment of Diffuse Cutaneous Mastocytosis. J. Pediatr. 2013, 162, 205–207. [Google Scholar] [CrossRef]
- Kristensen, T.; Vestergaard, H.; Bindslev-Jensen, C.; Mortz, C.G.; Kjaer, H.F.; Ollert, M.; Møller, M.B.; Broesby-Olsen, S. Prospective Evaluation of the Diagnostic Value of Sensitive KIT D816V Mutation Analysis of Blood in Adults with Suspected Systemic Mastocytosis. Allergy 2017, 72, 1737–1743. [Google Scholar] [CrossRef]
- Carter, M.C.; Bai, Y.; Ruiz-Esteves, K.N.; Scott, L.M.; Cantave, D.; Bolan, H.; Eisch, R.; Sun, X.; Hahn, J.; Maric, I.; et al. Detection of KIT D816V in Peripheral Blood of Children with Manifestations of Cutaneous Mastocytosis Suggests Systemic Disease. Br. J. Dermatol. 2018, 183, 775–782. [Google Scholar] [CrossRef]
- Czarny, J.; Żuk, M.; Żawrocki, A.; Plata-Nazar, K.; Biernat, W.; Niedoszytko, M.; Ługowska-Umer, H.; Nedoszytko, B.; Wasąg, B.; Nowicki, R.; et al. New Approach to Paediatric Mastocytosis: Implications of KIT D816V Mutation Detection in Peripheral Blood. Acta Derm. Venereol. 2020, 100, adv00149. [Google Scholar] [CrossRef]
- Wang, H.J.; Lin, Z.M.; Zhang, J.; Yin, J.H.; Yang, Y. A New Germline Mutation in KIT Associated with Diffuse Cutaneous Mastocytosis in a Chinese Family. Clin. Exp. Dermatol. 2014, 39, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Boxer, M.; Drummond, A.; Ogston, P.; Hodgins, M.; Burden, A.D. A Germline Mutation in KIT in Familial Diffuse Cutaneous Mastocytosis. J. Med. Genet. 2004, 41, e88. [Google Scholar] [CrossRef] [PubMed]
- Wangberg, H.; Willis, M.J.H.; Lindsey, D.; Schmidgal, E.C.; White, A.A. A Familial Case of Diffuse Cutaneous Mastocytosis. J. Allergy Clin. Immunol. Pract. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach–Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel Germline Mutation of KIT Associated With Familial Gastrointestinal Stromal Tumors and Mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.; Fiebig, B.; Lundberg, P.; Jaspers, N.-I.; Holzapfel, B.; Ghadimi, M.P.H.; Drebber, U.; Tuchscherer, A.; Ullrich, R.; Hartmann, K.; et al. Detection of the Germline KIT S476I Mutation in a Kindred with Familial Mastocytosis Associated with Gastrointestinal Stromal Tumors. J. Allergy Clin. Immunol. Pract. 2021, 9, 2123–2125.e1. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Kazi, J.U.; Zhao, H.; Sun, J. Germline Mutations of KIT in Gastrointestinal Stromal Tumor (GIST) and Mastocytosis. Cell Biosci. 2016, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Otani, I.M.; Carroll, R.W.; Yager, P.; Kroshinsky, D.; Murphy, S.; Hornick, J.L.; Akin, C.; Castells, M.; Walter, J.E. Diffuse Cutaneous Mastocytosis with Novel Somatic KIT Mutation K509I and Association with Tuberous Sclerosis. Clin. Case Rep. 2018, 6, 1834–1840. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and Standardization in Mastocytosis: Consensus Statements on Diagnostics, Treatment Recommendations and Response Criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef]
- Swarnkar, B.; Sarkar, R. Childhood Cutaneous Mastocytosis: Revisited. Indian J. Dermatol. 2023, 68, 121. [Google Scholar] [CrossRef]
- Özdemir, Ö. Cutaneous Mastocytosis in Childhood: An Update from the Literature. J. Clin. Pract. Res. 2023, 45, 311–320. [Google Scholar] [CrossRef]
- Nemat, K.; Abraham, S. Cutaneous Mastocytosis in Childhood. Allergol. Select 2022, 6, 1–10. [Google Scholar] [CrossRef]
- Lange, M.; Niedoszytko, M.; Renke, J.; Gleń, J.; Nedoszytko, B. Clinical Aspects of Paediatric Mastocytosis: A Review of 101 Cases. J. Eur. Acad. Dermatol. Venereol. 2011, 27, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Sandru, F.; Petca, R.C.; Costescu, M.; Dumitrașcu, M.C.; Popa, A.; Petca, A.; Miulescu, R.G. Cutaneous Mastocytosis in Childhood—Update from the Literature. J. Clin. Med. 2021, 10, 1474. [Google Scholar] [CrossRef]
- Carter, M.C.; Clayton, S.T.; Komarow, H.D.; Brittain, E.H.; Scott, L.M.; Cantave, D.; Gaskins, D.M.; Maric, I.; Metcalfe, D.D. Assessment of Clinical Findings, Tryptase Levels, and Bone Marrow Histopathology in the Management of Pediatric Mastocytosis. J. Allergy Clin. Immunol. 2015, 136, 1673–1679.e3. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Zawadzka, A.; Schrörs, S.; Słomka, J.; Ługowska-Umer, H.; Nedoszytko, B.; Nowicki, R. The Role of Serum Tryptase in the Diagnosis and Monitoring of Pediatric Mastocytosis: A Single-Center Experience. Adv. Dermatol. Alergol. 2017, 4, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Seth, N.; Tuano, K.T.; Buheis, M.; Chan, A.; Chinen, J. Serum Tryptase Levels in Pediatric Mastocytosis and Association with Systemic Symptoms. Ann. Allergy Asthma Immunol. 2020, 125, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K.; Plata-Nazar, K.; Lange, M.; Nedoszytko, B.; Niedoszytko, M.; Valent, P. Mediator-Related Symptoms and Anaphylaxis in Children with Mastocytosis. Int. J. Mol. Sci. 2021, 22, 2684. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; Vañõ-Galván, S.; Sánchez-Muñoz, L.; Morgado, J.M.; Matito, A.; Torrelo, A.; Jaén, P.; Schwartz, L.B.; Orfao, A.; Escribano, L. Increased Serum Baseline Tryptase Levels and Extensive Skin Involvement Are Predictors for the Severity of Mast Cell Activation Episodes in Children with Mastocytosis. Allergy 2012, 67, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.; Van, L.; DeLong, L.; Lawley, L.P. Severity of Cutaneous Findings Predict the Presence of Systemic Symptoms in Pediatric Maculopapular Cutaneous Mastocytosis. Pediatr. Dermatol. 2014, 31, 271–275. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Carter, M.; Kjaer, H.F.; Mortz, C.G.; Møller, M.B.; Kristensen, T.K.; Bindslev-Jensen, C.; Agertoft, L. Pediatric Expression of Mast Cell Activation Disorders. Immunol. Allergy Clin. N. Am. 2018, 38, 365–377. [Google Scholar] [CrossRef]
- Wiechers, T.; Rabenhorst, A.; Schick, T.; Preussner, L.M.; Förster, A.; Valent, P.; Horny, H.-P.; Sotlar, K.; Hartmann, K. Large Maculopapular Cutaneous Lesions Are Associated with Favorable Outcome in Childhood-Onset Mastocytosis. J. Allergy Clin. Immunol. 2015, 136, 1581–1590.e3. [Google Scholar] [CrossRef]
- Heide, R.; Zuidema, E.; Beishuizen, A.; Den Hollander, J.C.; Van Gysel, D.; Seyger, M.M.B.; Pasmans, S.G.M.A.; Kakourou, T.; Oranje, A.P. Clinical Aspects of Diffuse Cutaneous Mastocytosis in Children: Two Variants. Dermatology 2009, 219, 309–315. [Google Scholar] [CrossRef]
- Hosking, A.M.; Makdisi, J.; Ortenzio, F.; de Feraudy, S.; Smith, J.; Linden, K. Diffuse Cutaneous Mastocytosis: Case Report and Literature Review. Pediatr. Dermatol. 2018, 35, e348–e352. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Niedoszytko, M.; Nedoszytko, B.; Łata, J.; Trzeciak, M.; Biernat, W. Diffuse Cutaneous Mastocytosis: Analysis of 10 Cases and a Brief Review of the Literature. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Hannaford, R.; Rogers, M. Presentation of Cutaneous Mastocytosis in 173 Children. Australas. J. Dermatol. 2001, 42, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, J.M.; Cabral, C.A.S.; Lellis, R.F.; Ravelli, F.N. Bullous Congenital Diffuse Cutaneous Mastocytosis. An. Bras. Dermatol. 2020, 95, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Kleewein, K.; Lang, R.; Diem, A.; Vogel, T.; Pohla-Gubo, G.; Bauer, J.W.; Hintner, H.; Laimer, M. Diffuse Cutaneous Mastocytosis Masquerading as Epidermolysis Bullosa. Pediatr. Dermatol. 2011, 28, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Matito, A.; Azaña, J.M.; Torrelo, A.; Alvarez-Twose, I. Cutaneous Mastocytosis in Adults and Children. Immunol. Allergy Clin. N. Am. 2018, 38, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Olteanu, E.-G.; Bataneant, M.; Puiu, M.; Chirita-Emandi, A. When Mast Cells Run Amok: A Comprehensive Review and Case Study on Severe Neonatal Diffuse Cutaneous Mastocytosis. Genes 2023, 14, 2021. [Google Scholar] [CrossRef]
- Waxtein, L.M.; Elisa Vega-Memije, M.; Cortés-Franco, R.; Dominguez-Soto, L. Diffuse Cutaneous Mastocytosis with Bone Marrow Infiltration in a Child: A Case Report. Pediatr. Dermatol. 2000, 17, 198–201. [Google Scholar] [CrossRef]
- Angus, J.; Leach, I.H.; Grant, J.; Ravenscroft, J.C. Systemic Mastocytosis with Diffuse Cutaneous Involvement and Haematological Disease Presenting in Utero Treated Unsuccessfully with Vincristine. Clin. Exp. Dermatol. 2008, 33, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Walsh, D.; Drumm, B.; Watson, R. Bullous Mastocytosis: A Fatal Outcome. Pediatr. Dermatol. 1999, 16, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, M.; Ghanadan, A.; Jesri, S.B.; Sotudeh, S.; Ramyar, A. Diffuse Cutaneous Mastocytosis: Report of a Severe Case with Fatal Outcome. Dermatol. Online J. 2011, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.C.; Lam, J.M.; Leong, K.F. Childhood Solitary Cutaneous Mastocytoma: Clinical Manifestations, Diagnosis, Evaluation, and Management. Curr. Pediatr. Rev. 2019, 15, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Nair, B.; Sonthalia, S.; Aggarwal, I. Solitary Mastocytoma with Positive Darier′s Sign. Indian Dermatol. Online J. 2016, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Gopal, D.; Puri, P.; Singh, A.; Ramesh, V. Asymptomatic Solitary Cutaneous Mastocytoma: A Rare Presentation. Indian Dermatol. Online J. 2014, 59, 634. [Google Scholar] [CrossRef]
- Gülen, T. A Puzzling Mast Cell Trilogy: Anaphylaxis, MCAS, and Mastocytosis. Diagnostics 2023, 13, 3307. [Google Scholar] [CrossRef] [PubMed]
- Broesby-Olsen, S.; Dybedal, I.; Gülen, T.; Kristensen, T.; Møller, M.; Ackermann, L.; Sääf, M.; Karlsson, M.; Agertoft, L.; Brixen, K.; et al. Multidisciplinary Management of Mastocytosis: Nordic Expert Group Consensus. Acta Derm. Venereol. 2016, 96, 602–612. [Google Scholar] [CrossRef]
- Hoermann, G.; Cerny-Reiterer, S.; Perné, A.; Klauser, M.; Hoetzenecker, K.; Klein, K.; Müllauer, L.; Gröger, M.; Nijman, S.M.B.; Klepetko, W.; et al. Identification of Oncostatin M as a STAT5-Dependent Mediator of Bone Marrow Remodeling in KIT D816V-Positive Systemic Mastocytosis. Am. J. Pathol. 2011, 178, 2344–2356. [Google Scholar] [CrossRef]
- Greiner, G.; Witzeneder, N.; Berger, A.; Schmetterer, K.; Eisenwort, G.; Schiefer, A.-I.; Roos, S.; Popow-Kraupp, T.; Müllauer, L.; Zuber, J.; et al. CCL2 Is a KIT D816V–Dependent Modulator of the Bone Marrow Microenvironment in Systemic Mastocytosis. Blood 2017, 129, 371–382. [Google Scholar] [CrossRef]
- Ługowska-Umer, H.; Zabłotna, M.; Lange, M.; Niedoszytko, M.; Nowicki, R.; Nedoszytko, B. -2518A/G Polymorphism of Monocyte Chemotactic Protein 1 (MCP-1/CCL2) Is Associated with Cutaneous Mastocytosis. Adv. Dermatol. Alergol. 2021, 38, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K.; Ring, J.; Alvarez-Twose, I.; Orfao, A.; Escribano, L. Extensive Blistering Is a Predictor for Severe Complications in Children with Mastocytosis. Allergy 2012, 67, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Matito, A.; Carter, M. Cutaneous and Systemic Mastocytosis in Children: A Risk Factor for Anaphylaxis? Curr. Allergy Asthma Rep. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.; Camargo, C.A.; Bohlke, K.; Jick, H.; Miller, R.L.; Sheikh, A.; Simons, F.E.R. Epidemiology of Anaphylaxis: Findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann. Allergy Asthma Immunol. 2006, 97, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K.; Jofer, C.; Behrendt, H.; Ring, J. Anaphylaxis in Patients with Mastocytosis: A Study on History, Clinical Features and Risk Factors in 120 Patients. Allergy 2008, 63, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Heinze, A.; Kuemmet, T.J.; Chiu, Y.E.; Galbraith, S.S. Longitudinal Study of Pediatric Urticaria Pigmentosa. Pediatr. Dermatol. 2017, 34, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Jarkvist, J.; Brockow, K.; Gülen, T. Low Frequency of IgE-Mediated Food Hypersensitivity in Mastocytosis. J. Allergy Clin. Immunol. Pract. 2020, 8, 3093–3101. [Google Scholar] [CrossRef]
- Matito, A.; Morgado, J.M.; Sánchez-López, P.; Álvarez-Twose, I.; Sánchez-Muñoz, L.; Orfao, A.; Escribano, L. Management of Anesthesia in Adult and Pediatric Mastocytosis: A Study of the Spanish Network on Mastocytosis (REMA) Based on 726 Anesthetic Procedures. Int. Arch. Allergy Immunol. 2015, 167, 47–56. [Google Scholar] [CrossRef]
- Hussain, S.H. Pediatric Mastocytosis. Curr. Opin. Pediatr. 2020, 32, 531–538. [Google Scholar] [CrossRef]
- Lyons, J.J.; Yu, X.; Hughes, J.D.; Le, Q.T.; Jamil, A.; Bai, Y.; Ho, N.; Zhao, M.; Liu, Y.; O’Connell, M.P.; et al. Elevated Basal Serum Tryptase Identifies a Multisystem Disorder Associated with Increased TPSAB1 Copy Number. Nat. Genet. 2016, 48, 1564–1569. [Google Scholar] [CrossRef]
- Lyons, J.J. Hereditary Alpha Tryptasemia. Immunol. Allergy Clin. N. Am. 2018, 38, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.C.; Carter, M.C.; Korošec, P.; Bonadonna, P.; Schwartz, L.B.; Milner, J.D.; Caughey, G.H.; Metcalfe, D.D.; Lyons, J.J. Clinical Relevance of Inherited Genetic Differences in Human Tryptases. Ann. Allergy Asthma Immunol. 2021, 127, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Greiner, G.; Sprinzl, B.; Górska, A.; Ratzinger, F.; Gurbisz, M.; Witzeneder, N.; Schmetterer, K.G.; Gisslinger, B.; Uyanik, G.; Hadzijusufovic, E.; et al. Hereditary α Tryptasemia Is a Valid Genetic Biomarker for Severe Mediator-Related Symptoms in Mastocytosis. Blood 2021, 137, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, C.; Milner, J.D.; Lyons, J.J.; Vadas, P. Usefulness of Testing for Hereditary Alpha Tryptasemia in Symptomatic Patients with Elevated Tryptase. J. Allergy Clin. Immunol. Pract. 2020, 8, 2066–2067. [Google Scholar] [CrossRef] [PubMed]
- Zama, D.; Muratore, E.; Giannetti, A.; Neri, I.; Conti, F.; Magini, P.; Ferrari, S.; Pession, A. Case Report: Hereditary Alpha Tryptasemia in Children: A Pediatric Case Series and a Brief Overview of Literature. Front. Pediatr. 2021, 9, 716786. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Ługowska-Umer, H.; Niedoszytko, M.; Wasąg, B.; Limon, J.; Żawrocki, A.; Nedoszytko, B.; Sobjanek, M.; Plata-Nazar, K.; Nowicki, R. Diagnosis of Mastocytosis in Children and Adults in Daily Clinical Practice. Acta Derm. Venereol. 2016, 96, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, J.; Horny, H.P.; Kristensen, T.; Broesby-Olsen, S.; Zink, A.; Biedermann, T.; Brockow, K. Validation of Dermatopathological Criteria to Diagnose Cutaneous Lesions of Mastocytosis: Importance of KIT D816V Mutation Analysis. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1367–1375. [Google Scholar] [CrossRef]
- Greenberger, S.; Landov, H.; Confino, Y.; Vaknine, H.; Avivi, C.; Baum, S.; Barzilai, A. Immunophenotype of Pediatric-onset Mastocytosis Does Not Correlate with Clinical Course. Pediatr. Dermatol. 2019, 36, 477–481. [Google Scholar] [CrossRef]
- Morgado, J.M.; Sánchez-Muñoz, L.; Matito, A.; Mollejo, M.; Escribano, L.; Alvarez-Twose, I. Patterns of Expression of CD25 and CD30 on Skin Mast Cells in Pediatric Mastocytosis. J. Contemp. Immunol. 2014, 1, 46–56. [Google Scholar] [CrossRef]
- Hermans, M.A.W.; Pasmans, S.G.M.A.; Arends, N.J.T.; van den Bosch, T.P.P.; van Daele, P.L.A.; van Doorn, M.B.A.; Huisman, E.J.; Mooyaart, A.L.; Damman, J. Histopathological Characteristics Are Instrumental to Distinguish Monomorphic from Polymorphic Maculopapular Cutaneous Mastocytosis in Children. Clin. Exp. Dermatol. 2022, 47, 1694–1702. [Google Scholar] [CrossRef]
- Kristensen, T.; Vestergaard, H.; Møller, M.B. Improved Detection of the KIT D816V Mutation in Patients with Systemic Mastocytosis Using a Quantitative and Highly Sensitive Real-Time QPCR Assay. J. Mol. Diagn. 2011, 13, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Hoermann, G.; Bonadonna, P.; Hartmann, K.; Sperr, W.R.; Broesby-Olsen, S.; Brockow, K.; Niedoszytko, M.; Hermine, O.; Chantran, Y.; et al. The Normal Range of Baseline Tryptase Should Be 1 to 15 Ng/ML and Covers Healthy Individuals with HαT. J. Allergy Clin. Immunol. Pract. 2023, 11, 3010–3020. [Google Scholar] [CrossRef]
- Waters, A.M.; Park, H.J.; Weskamp, A.L.; Mateja, A.; Kachur, M.E.; Lyons, J.J.; Rosen, B.J.; Boggs, N.A. Elevated Basal Serum Tryptase: Disease Distribution and Variability in a Regional Health System. J. Allergy Clin. Immunol. Pract. 2022, 10, 2424–2435.e5. [Google Scholar] [CrossRef] [PubMed]
- Jesky, M.D.; Stringer, S.J.; Fenton, A.; Ng, K.P.; Yadav, P.; Ndumbo, M.; McCann, K.; Plant, T.; Dasgupta, I.; Harding, S.J.; et al. Serum Tryptase Concentration and Progression to End-stage Renal Disease. Eur. J. Clin. Investig. 2016, 46, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Rama, T.A.; Castells, M. Triggers of Anaphylaxis in Mastocytosis Patients: Evidence of the Current Drug-Avoidance Recommendation. Curr. Treat. Options Pediatr. 2023. [Google Scholar] [CrossRef]
- Robey, R.C.; Wilcock, A.; Bonin, H.; Beaman, G.; Myers, B.; Grattan, C.; Briggs, T.A.; Arkwright, P.D. Hereditary Alpha-Tryptasemia: UK Prevalence and Variability in Disease Expression. J. Allergy Clin. Immunol. Pract. 2020, 8, 3549–3556. [Google Scholar] [CrossRef] [PubMed]
- Kačar, M.; Rijavec, M.; Šelb, J.; Korošec, P. Clonal Mast Cell Disorders and Hereditary A-tryptasemia as Risk Factors for Anaphylaxis. Clin. Exp. Allergy 2023, 53, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Sordi, B.; Vanderwert, F.; Crupi, F.; Gesullo, F.; Zanotti, R.; Bonadonna, P.; Crosera, L.; Elena, C.; Fiorelli, N.; Ferrari, J.; et al. Disease Correlates and Clinical Relevance of Hereditary α-Tryptasemia in Patients with Systemic Mastocytosis. J. Allergy Clin. Immunol. 2023, 151, 485–493.e11. [Google Scholar] [CrossRef]
- Bernett, C.N.; Mammino, J.J. Cutaneous Leiomyomas; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lang, K.; Reifenberger, J.; Ruzicka, T.; Megahed, M. Type 1 Segmental Cutaneous Leiomyomatosis. Clin. Exp. Dermatol. 2002, 27, 649–650. [Google Scholar] [CrossRef]
- Jezierska, M.; Stefanowicz, J.; Romanowicz, G.; Kosiak, W.; Lange, M. Langerhans Cell Histiocytosis in Children—A Disease with Many Faces. Recent Advances in Pathogenesis, Diagnostic Examinations and Treatment. Adv. Dermatol. Alergol. 2018, 35, 6–17. [Google Scholar] [CrossRef]
- Brazel, M.; Desai, A.; Are, A.; Motaparthi, K. Staphylococcal Scalded Skin Syndrome and Bullous Impetigo. Medicina 2021, 57, 1157. [Google Scholar] [CrossRef]
- Rayinda, T.; Mira Oktarina, D.A.; Danarti, R. Diffuse Cutaneous Mastocytosis Masquerading as Linear IgA Bullous Dermatosis of Childhood. Dermatol. Rep. 2021, 13, 9021. [Google Scholar] [CrossRef] [PubMed]
- Lammer, J.; Hein, R.; Roenneberg, S.; Biedermann, T.; Volz, T. Drug-Induced Linear IgA Bullous Dermatosis: A Case Report and Review of the Literature. Acta Derm. Venereol. 2019, 99, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Golitz, L.E.; Weston, W.L.; Lane, A.T. Bullous Mastocytosis: Diffuse Cutaneous Mastocytosis with Extensive Blisters Mimicking Scalded Skin Syndrome or Erythema Multiforme. Pediatr. Dermatol. 1984, 1, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Oranje, A.P.; Soekanto, W.; Sukardi, A.; Vuzevski, V.D.; van der Willigen, A.; Afiani, H.M. Diffuse Cutaneous Mastocytosis Mimicking Staphylococcal Scalded-Skin Syndrome: Report of Three Cases. Pediatr. Dermatol. 1991, 8, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Kokubo, T.; Akaishi, M.; Iida, K.; Korematsu, S. Neonatal Onset Diffuse Cutaneous Mastocytosis: A Case Report and Review of the Literature. Pediatr. Dermatol. 2011, 28, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Kunnath, M.; Minnaar, G. A Case of Non-Pigment Forming Cutaneous Mastocytosis, Masquerading as Infantile Eczema. Arch. Dis. Child. 2010, 95, A26–A27. [Google Scholar] [CrossRef]
- Bourji, L.; Kurban, M.; Abbas, O. Solitary Mastocytoma Mimicking Granuloma Faciale. Int. J. Dermatol. 2014, 53, e587–e588. [Google Scholar] [CrossRef]
- Gurnee, E.A.; Johansen, M.L.; Phung, T.L.; Guo, E.L.; Fong, A.; Tollefson, M.; Nguyen, H.; Brandling-Bennett, H.; Moriarty, N.; Paller, A.S.; et al. Pediatric Maculopapular Cutaneous Mastocytosis: Retrospective Review of Signs, Symptoms, and Associated Conditions. Pediatr. Dermatol. 2021, 38, 159–163. [Google Scholar] [CrossRef]
- Czarny, J.; Renke, J.; Żawrocki, A.; Nowicki, R.J.; Lange, M. Natural Evolution in Pediatric Cutaneous Mastocytosis: 10-year Follow-up. Int. J. Dermatol. 2021, 60, 1253–1257. [Google Scholar] [CrossRef]
- Le, M.; Miedzybrodzki, B.; Olynych, T.; Chapdelaine, H.; Ben-Shoshan, M. Natural History and Treatment of Cutaneous and Systemic Mastocytosis. Postgrad. Med. 2017, 129, 896–901. [Google Scholar] [CrossRef]
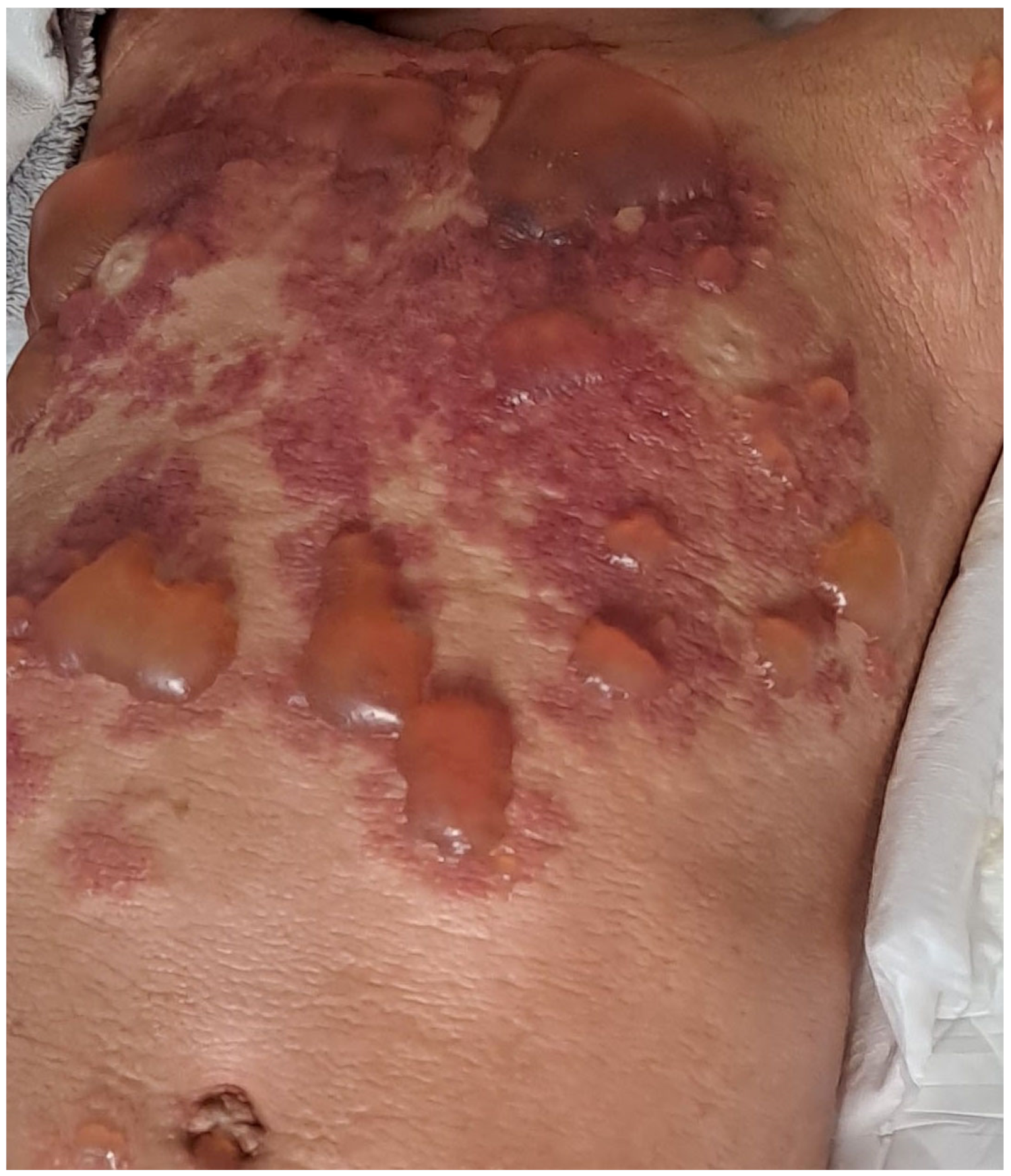
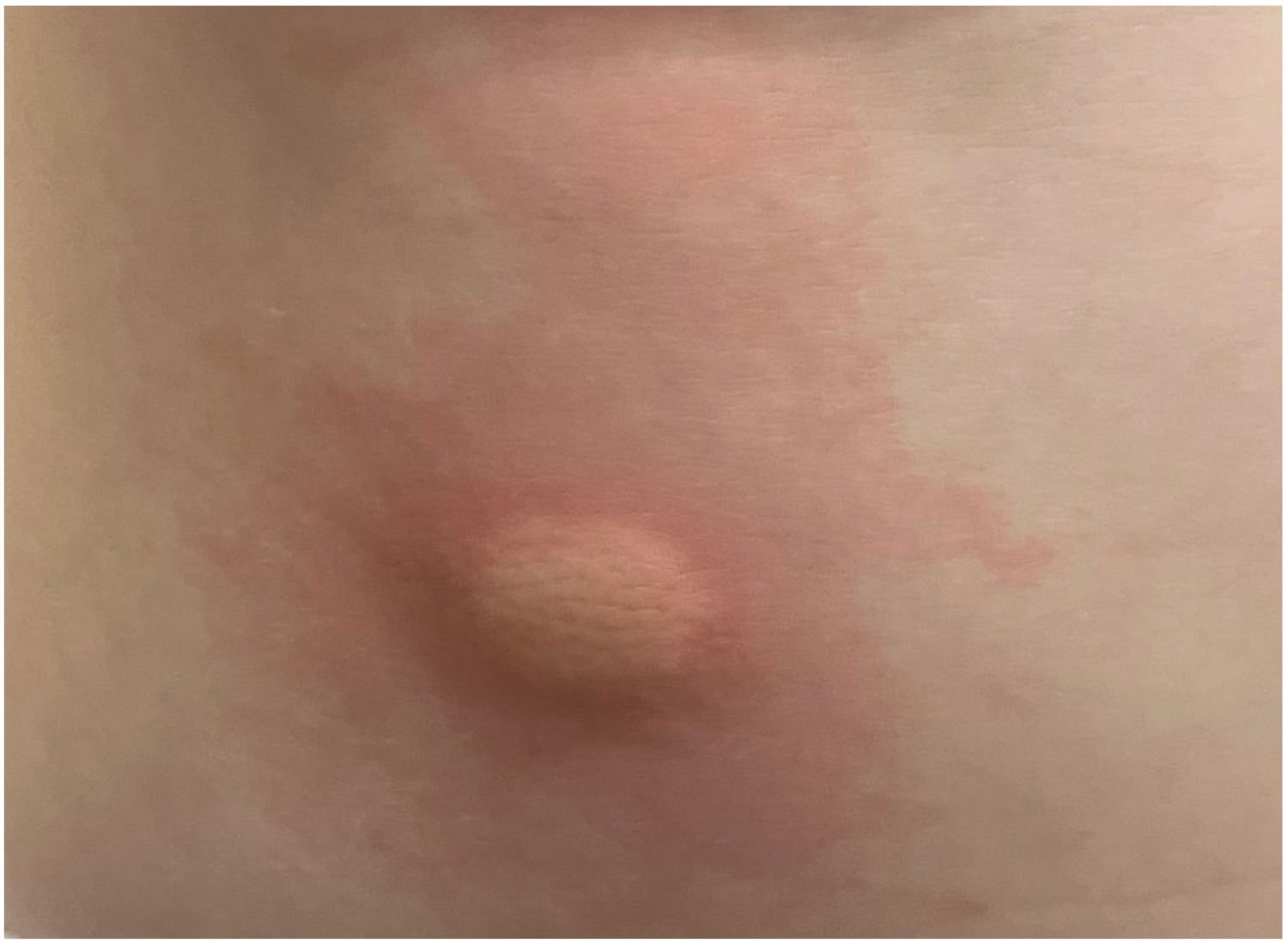

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cutaneous mastocytosis (CM)
|
| Systemic mastocytosis (SM) Nonadvanced forms of SM
|
| Mast cell sarcoma (MCS) |
| Diagnostic Procedure | Main Indications | Comments |
|---|---|---|
| Medical History | All children with suspected CM |
|
| Physical Examination | All children with suspected CM |
|
| Elicitation of Darier’s sign | All children with suspected CM |
|
| PB count with differential | All children with suspected CM |
|
| Biochemistry | All children with suspected CM |
|
| Basal serum tryptase level | All children with CM |
|
| Abdominal ultrasound/CT | All children with CM | Hepatosplenomegaly and lymphadenopathy are strong indicators of SM |
| Skin biopsy and histological examination of lesional skin | Children with suspected CM in whom:
| The use of antibodies against tryptase and/or CD117 are recommended immunohistochemical markers of MCs |
| KIT D816V mutation in lesional skin (optionally other activating KIT mutations) | Children with suspected CM in whom:
|
|
| KIT D816V mutation in PB (optionally other activating KIT mutations) | Children with suspected SM | The determination of activating KIT mutation(s) is recommended for risk stratification of SM before deciding on a BM biopsy |
| BM biopsy | Children with suspected SM | BM biopsy should be considered in children with:
|
| Genetic test for TPSAB1 copy number | Children with:
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ługowska-Umer, H.; Czarny, J.; Rydz, A.; Nowicki, R.J.; Lange, M. Current Challenges in the Diagnosis of Pediatric Cutaneous Mastocytosis. Diagnostics 2023, 13, 3583. https://doi.org/10.3390/diagnostics13233583
Ługowska-Umer H, Czarny J, Rydz A, Nowicki RJ, Lange M. Current Challenges in the Diagnosis of Pediatric Cutaneous Mastocytosis. Diagnostics. 2023; 13(23):3583. https://doi.org/10.3390/diagnostics13233583
Chicago/Turabian StyleŁugowska-Umer, Hanna, Justyna Czarny, Agnieszka Rydz, Roman J. Nowicki, and Magdalena Lange. 2023. "Current Challenges in the Diagnosis of Pediatric Cutaneous Mastocytosis" Diagnostics 13, no. 23: 3583. https://doi.org/10.3390/diagnostics13233583
APA StyleŁugowska-Umer, H., Czarny, J., Rydz, A., Nowicki, R. J., & Lange, M. (2023). Current Challenges in the Diagnosis of Pediatric Cutaneous Mastocytosis. Diagnostics, 13(23), 3583. https://doi.org/10.3390/diagnostics13233583