
The Role of the Spleen and the Place of Splenectomy in Autoimmune Hemolytic Anemia—A Review of Current Knowledge

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
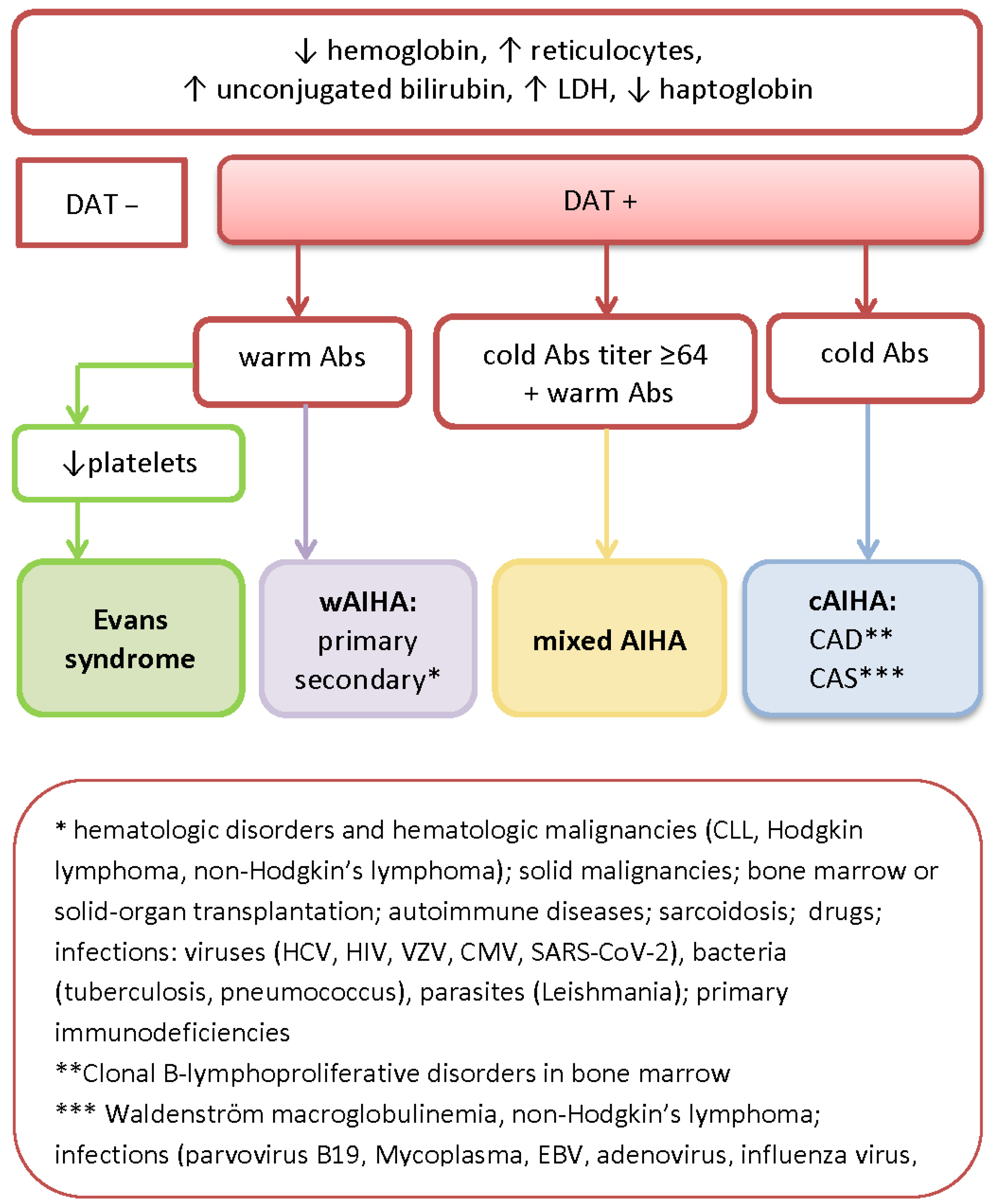
2. Diagnostic Workup, Serological Features, and the Classification of AIHA
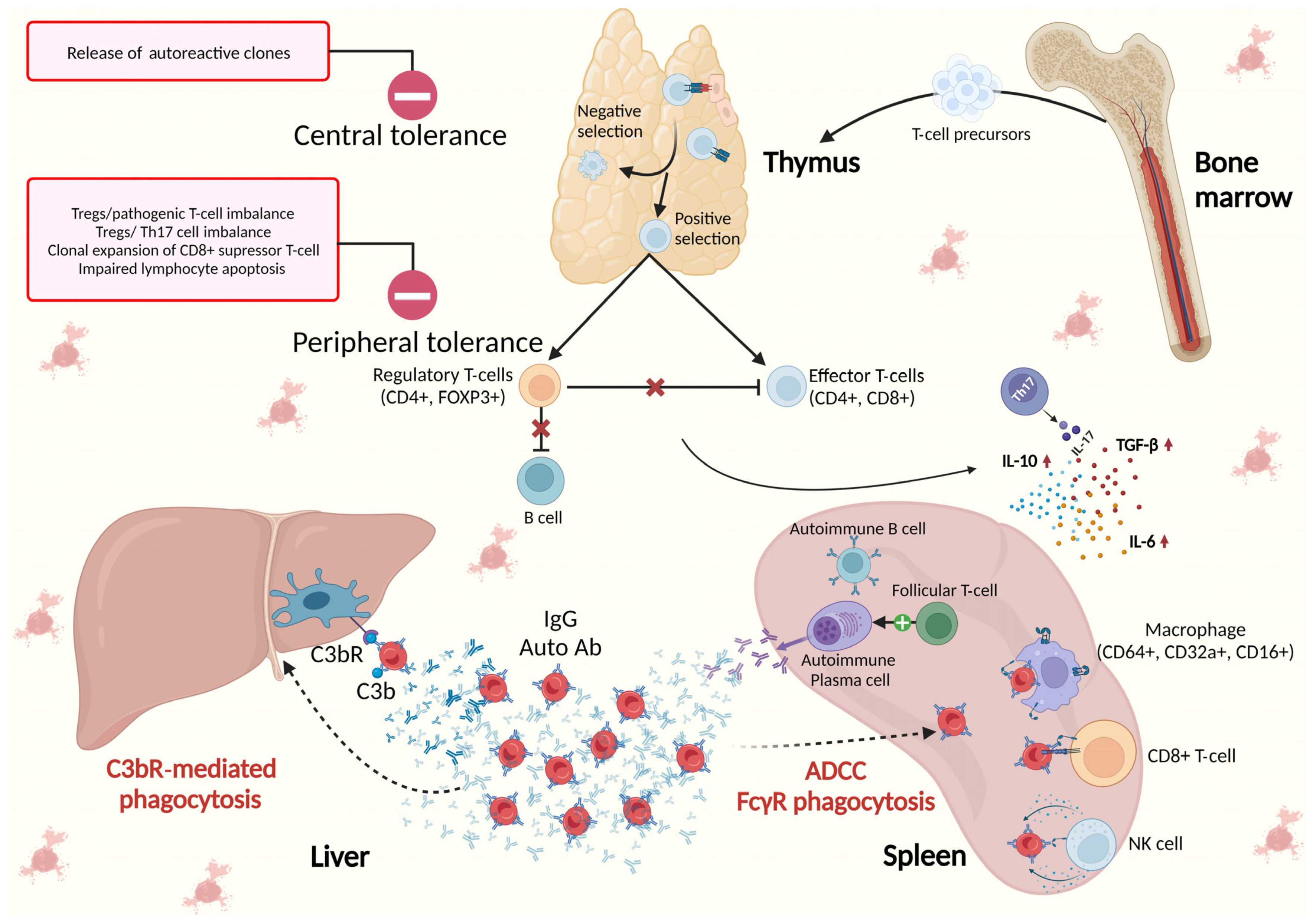
3. The Role of the Spleen and Bone Marrow in the Immunological Background of AIHA
3.1. Spleen as the Place of Origin of Autoantibodies in AIHA
3.2. Immune Clearance of Autoantibodies by Spleen Macrophages
3.3. Autoimmune Reaction against Bone Marrow and Extramedullary Hematopoiesis in the Spleen
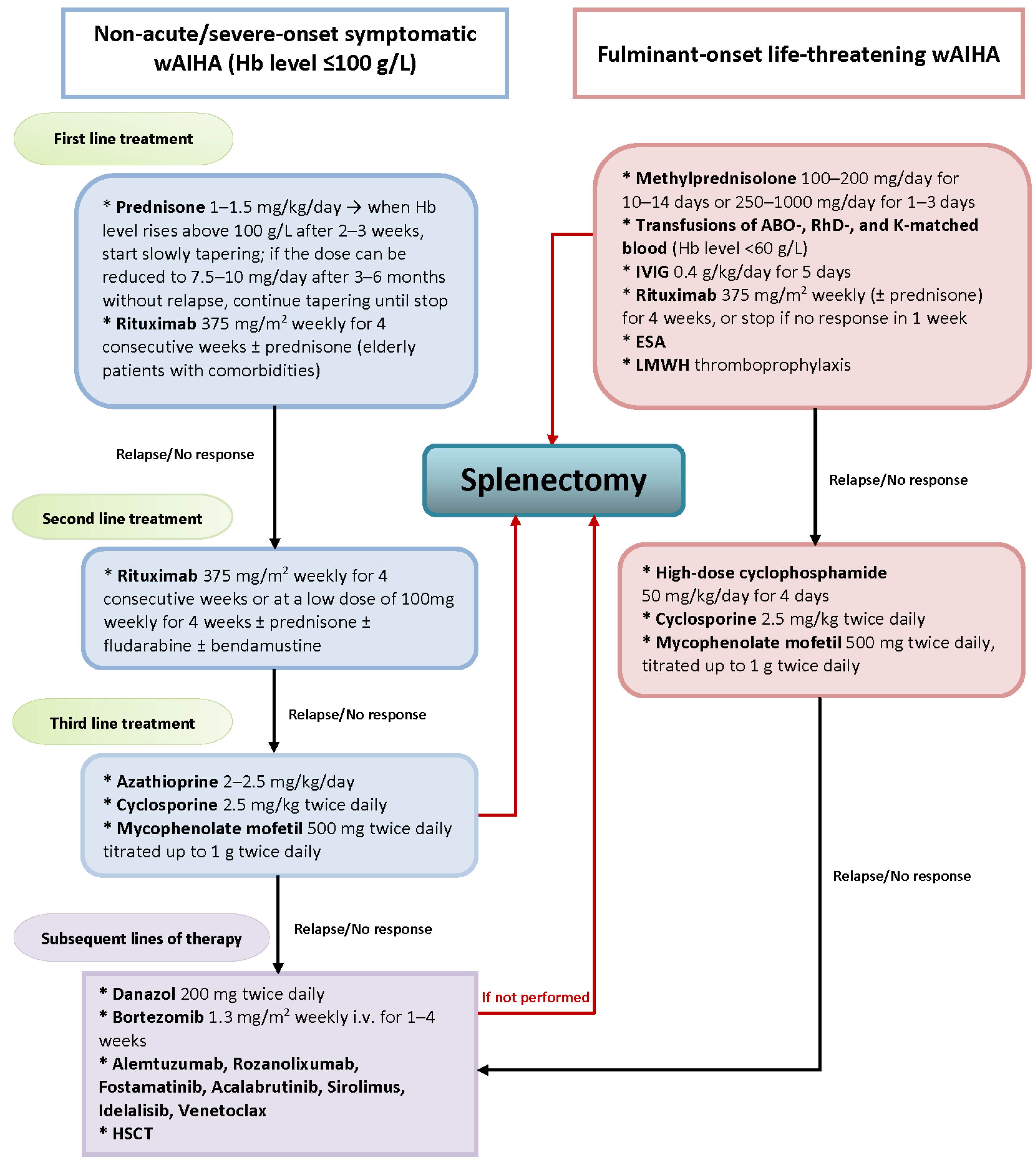
4. Novel Treatment Strategies for AIHA
5. The Place of Splenectomy in the Era of Novel AIHA Treatment
6. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berentsen, S.; Barcellini, W. Autoimmune Hemolytic Anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef]
- Barcellini, W.; Zaninoni, A.; Giannotta, J.A.; Fattizzo, B. New Insights in Autoimmune Hemolytic Anemia: From Pathogenesis to Therapy Stage 1. J. Clin. Med. 2020, 9, 3859. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A.; British Society for Haematology. The diagnosis and management of primary autoimmune haemolyticanaemia. Br. J. Haematol. 2017, 176, 395–411. [Google Scholar] [CrossRef]
- Kuter, D.J. Warm autoimmune hemolytic anemia and the best treatment strategies. Hematology 2022, 2022, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Jäger, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020, 41, 100648. [Google Scholar] [CrossRef]
- Jacobs, J.W.; Figueroa Villalba, C.A.; Booth, G.S.; Woo, J.S.; Stephens, L.D.; Adkins, B.D. Clinical and epidemiological features of paroxysmal cold hemoglobinuria: A systematic review. Blood Adv. 2023, 13, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Hill, A.; Berentsen, S. Defining autoimmune hemolytic anemia: A systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019, 3, 1897–1906. [Google Scholar] [CrossRef]
- Audia, S.; Grienay, N.; Mounier, M.; Michel, M.; Bonnotte, B. Evans’ Syndrome: From Diagnosis to Treatment. J. Clin. Med. 2020, 9, 3851. [Google Scholar] [CrossRef]
- Barcellini, W. Pitfalls in the diagnosis of autoimmune haemolytic anaemia. Blood Transfus. 2015, 13, 3–5. [Google Scholar]
- Zarandona, J.M.; Yazer, M.H. The role of the Coombs test in evaluating hemolysis in adults. CMAJ 2006, 174, 305–307. [Google Scholar] [CrossRef][Green Version]
- Ames, P.R.J.; Merashli, M.; Bucci, T.; Pastori, D.; Pignatelli, P.; Arcaro, A.; Gentile, F. Antiphospholipid Antibodies and Autoimmune Haemolytic Anaemia: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 4120. [Google Scholar] [CrossRef] [PubMed]
- Parker, V.; Tormey, C.A. The Direct Antiglobulin Test: Indications, Interpretation, and Pitfalls. Arch. Pathol. Lab. Med. 2017, 141, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Ribkoff, J.; Deloughery, T.; Olson, S.R. Incidence of False-Positive Direct Anti Globulin Tests (DAT): A Single-Center Retrospective Analysis. Blood 2020, 136, 6. [Google Scholar] [CrossRef]
- Michalak, S.S.; Olewicz-Gawlik, A.; Rupa-Matysek, J.; Wolny-Rokicka, E.; Nowakowska, E.; Gil, L. Autoimmune hemolytic anemia: Current knowledge and perspectives. Immun. Ageing 2020, 17, 38. [Google Scholar] [CrossRef]
- Fattizzo, B.; Michel, M.; Zaninoni, A.; Giannotta, J.; Guillet, S.; Frederiksen, H.; Vos, J.M.I.; Mauro, F.R.; Jilma, B.; Patriarca, A.; et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: A multicenter international study. Haematologica 2021, 106, 622–625. [Google Scholar] [CrossRef]
- Małecka, A.; Trøen, G.; Tierens, A.; Østlie, I.; Małecki, J.; Randen, U.; Wang, J.; Berentsen, S.; Tjønnfjord, G.E.; Delabie, J.M.A. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br. J. Haematol. 2018, 183, 838–842. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef]
- Pelletier, N.; McHeyzer-Williams, L.J.; Wong, K.A.; Urich, E.; Fazilleau, N.; McHeyzer-Williams, M.G. Plasma cells negatively regulate the follicular helper T cell program. Nat. Immunol. 2010, 11, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Slamanig, S.A.; Nolte, M.A. The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells 2021, 10, 1508. [Google Scholar] [CrossRef]
- Liu, X.; Yao, J.; Zhao, Y.; Wang, J.; Qi, H. Heterogeneous plasma cells and long-lived subsets in response to immunization, autoantigen and microbiota. Nat. Immunol. 2022, 23, 1564–1576. [Google Scholar] [CrossRef]
- Jang, E.; Cho, S.; Pyo, S.; Nam, J.W.; Youn, J. An Inflammatory Loop Between Spleen-Derived Myeloid Cells and CD4+ T Cells Leads to Accumulation of Long-Lived Plasma Cells That Exacerbates Lupus Autoimmunity. Front. Immunol. 2021, 12, 631472. [Google Scholar] [CrossRef]
- Hoyer, B.F.; Moser, K.; Hauser, A.E.; Peddinghaus, A.; Voigt, C.; Eilat, D.; Radbruch, A.; Hiepe, F.; Manz, R.A. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004, 199, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W. New Insights in the Pathogenesis of Autoimmune Hemolytic Anemia. Transfus. Med. Hemotherapy 2015, 42, 287–293. [Google Scholar] [CrossRef]
- Porpaczy, E.; Jäger, U. How I manage autoimmune cytopenias in patients with lymphoid cancer. Blood 2022, 139, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Khanmohammadi, S.; Shabani, M.; Tabary, M.; Rayzan, E.; Rezaei, N. Lymphoma in the setting of autoimmune diseases: A review of association and mechanisms. Crit. Rev. Oncol Hematol. 2020, 150, 102945. [Google Scholar] [CrossRef]
- Marques, H.S.; de Brito, B.B.; da Silva, F.A.F.; Santos, M.L.C.; de Souza, J.C.B.; Correia, T.M.L.; Lopes, L.W.; Neres, N.S.M.; Dórea, R.S.D.M.; Dantas, A.C.S.; et al. Relationship between Th17 immune response and cancer. World J. Clin. Oncol 2021, 12, 845–867. [Google Scholar] [CrossRef]
- Kanellopoulou, T. Autoimmune hemolytic anemia in solid organ transplantation-The role of immunosuppression. Clin. Transpl. 2017, 31, e13031. [Google Scholar] [CrossRef]
- Kennedy, C.; Jackson, D.E. The effect of HLA matching and donor relatedness on the risk of autoimmune haemolytic anaemia in haematopoietic stem cell transplant recipients: A systematic review and meta-analysis. EJHaem 2022, 3, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Maia, K.; Leguelinel-Blache, G.; Eiden, C.; Bernard, J.; Donnadieu-Rigole, H.; Peyrière, H. Drug-Induced Autoimmune Hemolytic Anemia: Detection of New Signals in the World Pharmacovigilance Database and Risk Assessment in a Nationwide Cohort Study in France. Blood 2022, 140, 8176–8177. [Google Scholar]
- Leaf, R.K.; Ferreri, C.; Rangachari, D.; Mier, J.; Witteles, W.; Ansstas, G.; Anagnostou, T.; Zubiri, L.; Piotrowska, Z.; Oo, T.H.; et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am. J. Hematol. 2019, 94, 563–574. [Google Scholar] [CrossRef]
- Cvetković, M.; Pantić, N.; Virijević, M.; Pravdić, Z.; Sabljić, N.; Mitrović, M.; Suvajdžić-Vuković, N. Relapse of Evans syndrome following BNT162b2 (Pfizer-BioNTech) COVID-19 vaccine: Case report and literature review. J. Infect. Dev. Ctries 2023, 17, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br. J. Haematol. 2017, 177, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wu, Y.; Guo, G.; Zeng, J.; Liu, Y.; Wu, Y. Piperacillin-tazobactam induced immune hemolytic anemia led to increased renal impairment and eventual death from multiple organ failure in a patient with hypertensive nephropathy: Case report and literature review. BMC Nephrol. 2023, 24, 173. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Parker, C.J.; Prchal, J.T. How Do Red Blood Cells Die? Front. Physiol. 2021, 12, 655393. [Google Scholar] [CrossRef]
- Kaufman, R. Red Blood Cell Life Span, Senescence, and destruction. In Anemia: Pathophysiology, Diagnosis and Management; Benz, E.J., Berliner, N., Schiffman, F.J., Eds.; Cambridge University Press: Cambridge, UK, 2017; pp. 19–22. [Google Scholar]
- Klei, T.R.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef]
- Ahrens, N.; Pagenkopf, C.; Kiesewetter, H.; Salama, A. CD47 is expressed at normal levels in patients with autoimmune haemolytic anaemia and/or immune thrombocytopenia. Transfus. Med. 2006, 16, 397–402. [Google Scholar] [CrossRef]
- Willekens, F.L.; Roerdinkholder-Stoelwinder, B.; Groenen-Döpp, Y.A.; Bos, H.J.; Bosman, G.J.; van den Bos, A.G.; Verkleij, A.J.; Were, J.M. Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood 2003, 101, 747–751. [Google Scholar] [CrossRef]
- Ohmes, J.; Comdühr, S.; Akbarzadeh, R.; Riemekasten, G.; Humrich, J.Y. Dysregulation and chronicity of pathogenic T cell responses in the pre-diseased stage of lupus. Front. Immunol. 2022, 13, 1007078. [Google Scholar] [CrossRef]
- Aladjidi, N.; Leverger, G.; Leblanc, T.; Picat, M.Q.; Michel, G.; Bertrand, Y.; Bader-Meunier, B.; Robert, A.; Nelken, B.; Gandemer, V.; et al. New insights into childhood autoimmune hemolytic anemia: A French national observational study of 265 children. Haematologica 2011, 96, 655–663. [Google Scholar] [CrossRef]
- Arbach, O.; Funck, R.; Seibt, F.; Salama, A. Erythropoietin May Improve Anemia in Patients with Autoimmune Hemolytic Anemia Associated with Reticulocytopenia. Transfus. Med. Hemotherapy 2012, 39, 221–223. [Google Scholar] [CrossRef]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress Erythropoiesis is a Key Inflammatory Response. Cells 2020, 9, 634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, G.; Zhang, W. Effect of TLR4/MyD88 signaling pathway on sepsis-associated acute respiratory distress syndrome in rats, via regulation of macrophage activation and inflammatory response. Exp. Ther. Med. 2018, 15, 3376–3384. [Google Scholar] [CrossRef]
- Cuenca, A.G.; Joiner, D.N.; Gentile, L.F.; Cuenca, A.L.; Wynn, J.L.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Behrns, K.E.; Efron, P.A.; Nacionales, D.; et al. TRIF-dependent innate immune activation is critical for survival to neonatal gram-negative sepsis. J. Immunol. 2015, 194, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Kiripolsky, J.; Romano, R.A.; Kasperek, E.M.; Yu, G.; Kramer, J.M. Activation of Myd88-Dependent TLRs Mediates Local and Systemic Inflammation in a Mouse Model of Primary Sjögren’s Syndrome. Front. Immunol. 2020, 10, 2963. [Google Scholar] [CrossRef]
- Zhang, H.; Rodriguez, S.; Wang, L.; Wang, S.; Serezani, H.; Kapur, R.; Cardoso, A.A.; Carlesso, N. Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88. Stem Cell Rep. 2016, 6, 940–956. [Google Scholar] [CrossRef]
- Noel, J.G.; Ramser, S.W.; Pitstick, L.; Goetzman, H.S.; Dale, E.L.; Potter, A.; Adam, M.; Potter, S.S.; Gardner, J.C. IL-1/MyD88-Dependent G-CSF and IL-6 Secretion Mediates Postburn Anemia. J. Immunol. 2023, 210, 972–980. [Google Scholar] [CrossRef]
- Lauda, E.; Haam, E. Importance of the spleen as a reservoir for red blood cells. Exp Biol Med. 1931, 29, 260–262. [Google Scholar] [CrossRef]
- Coppin, E.; Florentin, J.; Vasamsetti, S.B.; Arunkumar, A.; Sembrat, J.; Rojas, M.; Dutta, P. Splenic hematopoietic stem cells display a pre-activated phenotype. Immunol. Cell Biol. 2018, 96, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, D.; Song, P.; Jiang, F.; Chi, X.; Zhang, T. Exposure to hypoxia causes stress erythropoiesis and downregulates immune response genes in spleen of mice. BMC Genom. 2021, 22, 413. [Google Scholar] [CrossRef] [PubMed]
- Despotovic, J.M.; Kim, T.O. Cold AIHA and the best treatment strategies. Hematology 2022, 2022, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, R.A. Warm Autoimmune Hemolytic Anemia. N. Engl. J. Med. 2019, 381, 647–654. [Google Scholar] [CrossRef]
- Berentsen, S.; Fattizzo, B.; Barcellini, W. The choice of new treatments in autoimmune hemolytic anemia: How to pick from the basket? Front. Immunol. 2023, 14, 1180509. [Google Scholar] [CrossRef] [PubMed]
- Roumier, M.; Loustau, V.; Guillaud, C.; Languille, L.; Mahevas, M.; Khellaf, M.; Limal, N.; Noizat-Pirenne, F.; Godeau, B.; Michel, M. Characteristics and outcome of warm autoimmune hemolytic anemia in adults: New insights based on a single-center experience with 60 patients. Am. J. Hematol. 2014, 89, E150–E155. [Google Scholar] [CrossRef]
- Reynaud, Q.; Durieu, I.; Dutertre, M.; Ledochowski, S.; Durupt, S.; Michallet, A.S.; Vital-Durand, D.; Lega, J.C. Efficacy and safety of rituximab in auto-immune hemolytic anemia: A meta-analysis of 21 studies. Autoimmun. Rev. 2015, 14, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A.; Radice, T.; Nichele, I.; Di Bona, E.; Lunghi, M.; Tassinari, C.; Alfinito, F.; Ferrari, A.; et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: A GIMEMA study of 308 patients. Blood 2014, 124, 2930–2936. [Google Scholar] [CrossRef]
- Dierickx, D.; Kentos, A.; Delannoy, A. The role of rituximab in adults with warm antibody autoimmune hemolytic anemia. Blood 2015, 125, 3223–3229. [Google Scholar] [CrossRef]
- Jomarrón, I.G.; Rubio, M.L.; Arias, M.M.; Arrizabalaga, B.; de la Iglesia, S.; Beneitez, D.; Sáez, M.I.; Cervera, A.; Recasens, V.; Herrera, A.; et al. Autoimmune haemolytic anaemias: A retrospective study of 93 patients. Med. Clin. 2020, 154, 331–337. [Google Scholar] [CrossRef]
- Barcellini, W.; Zaja, F.; Zaninoni, A.; Imperiali, F.G.; Di Bona, E.; Fattizzo, B.; Consonni, D.; Cortelezzi, A.; Zanella, A. Sustained response to low-dose rituximab in idiopathic autoimmune hemolytic anemia. Eur. J. Haematol. 2013, 91, 546–551. [Google Scholar] [CrossRef]
- Kuter, D.J.; Rogers, K.A.; Boxer, M.A.; Choi, M.; Agajanian, R.; Arnold, D.M.; Broome, C.M.; Field, J.J.; Murakhovskaya, I.; Numerof, R.; et al. Fostamatinib for the treatment of warm antibody autoimmune hemolytic anemia: Phase 2, multicenter, open-label study. Am. J. Hematol. 2022, 97, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Quinquenel, A.; Godet, S.; Dartigeas, C.; Ysebaert, L.; Dupuis, J.; Ohanyan, H.; Collignon, A.; Gilardin, L.; Lepretre, S.; Dilhuydy, M.S.; et al. Ibrutinib and idelalisib in the management of CLL-associated autoimmune cytopenias: A study from the FILO group. Am. J. Hematol. 2019, 94, E183–E185. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Ahn, I.E.; Sivina, M.; Ferrajoli, A.; Wierda, W.G.; Estrov, Z.; Konoplev, S.N.; Jain, N.; O’Brien, S.; Farooqui, M.; et al. Autoimmune cytopenias in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica 2016, 101, e254–e258. [Google Scholar] [CrossRef]
- Cavazzini, F.; Lista, E.; Quaglia, F.M.; Formigaro, L.; Cavallari, M.; Martinelli, S.; Rigolin, G.M.; Foà, R.; Cuneo, A. Response to ibrutinib of refractory life-threatening autoimmune hemolytic anemia occurring in a relapsed chronic lymphocytic leukemia patient with 17p deletion. Leuk. Lymphoma 2016, 57, 2685–2688. [Google Scholar] [CrossRef]
- Garcia-Horton, A.; Bernard, R.S.; Lazo-Langner, A.; Xenocostas, A.; Mangel, J.; Howson-Jan, K.; Lam, S.; Hsia, C.C. Safe Start of Ibrutinib in Patients with Chronic Lymphocytic Leukemia and Uncontrolled Autoimmune Hemolytic Anemia. Blood 2018, 132, 5560. [Google Scholar] [CrossRef]
- Montillo, M.; O’Brien, S.; Tedeschi, A.; Byrd, J.C.; Dearden, C.; Gill, D.; Brown, J.R.; Barrientos, J.C.; Mulligan, S.P.; Furman, R.R.; et al. Ibrutinib in previously treated chronic lymphocytic leukemia patients with autoimmune cytopenias in the resonate study. Blood Cancer J. 2017, 3, e524. [Google Scholar] [CrossRef]
- Galinier, A.; Delwail, V.; Puyade, M. Ibrutinib Is Effective in the Treatment of Autoimmune Haemolytic Anaemia in Mantle Cell Lymphoma. Case Rep. Oncol 2017, 10, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ji, J.; Du, Y.; Huang, Y.; Gu, H.; Chen, M.; Wu, R.; Han, B. Sirolimus is effectivefor primary relapsed/refractory autoimmune cytopenia: A multicenter study. Exp. Hematol. 2020, 89, 87–95. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. How I treat warm autoimmune hemolytic anemia. Blood 2021, 137, 1283–1294. [Google Scholar] [CrossRef]
- Fattizzo, B.; Barcellini, W. New Therapies for the Treatment of Warm Autoimmune Hemolytic Anemia. Transfus. Med. Rev. 2022, 36, 175–180. [Google Scholar] [CrossRef]
- Xiao, Z.; Murakhovskaya, I. Development of New Drugs for Autoimmune Hemolytic Anemia. Pharmaceutics 2022, 14, 1035. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Navarro, P.; Delgado-García, A.; Rodríguez-Gil, M.A.; Puerta-Puerta, J.M. Venetoclax for treating refractory autoimmune hemolytic anemia in chronic lymphocytic leukemia: Report of two cases in Spain. Haematologica 2023, 108, 2261–2264. [Google Scholar] [CrossRef]
- Yao, M.; Zhang, J.; Li, Y.; Lv, L.; Jia, L.; Yang, C.; Huang, Y.; Liu, H.; Wang, J.; Chen, M.; et al. Combination of low-dose rituximab, bortezomib and dexamethasone for the treatment of autoimmune hemolytic anemia. Medicine 2022, 101, e28679. [Google Scholar] [CrossRef] [PubMed]
- Ames, P.R.J.; Jeffrey, S. Bortezomib and rituximab in multiply relapsed primary warm autoimmune hemolytic anemia. Ann. Hematol. 2021, 100, 2415–2416. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhuang, J.; Yang, C.; Zhang, L.; Wang, W.; Cai, H.; Yu, Y.; Li, J.; Zhou, D.; Han, B. Rapid response to a single-dose rituximab combined with bortezomib in refractory and relapsed warm autoimmune hemolytic anemia. Ann. Hematol. 2020, 99, 1141–1143. [Google Scholar] [CrossRef]
- Chineke, I.; Kagbo-Kue, S.; Aniekwena, J.; Rose, M. Successful treatment of severe idiopathic mixed autoimmune hemolytic anemia with bortezomib and intravenous immunoglobulin. Int. J. Blood Transfus. Immunohematol. 2019, 9, 100046Z02IC2019. [Google Scholar] [CrossRef]
- Jain, A.; Gupta, D.K. Daratumumab for refractory warm autoimmune hemolytic anemia. Ann. Hematol. 2021, 100, 1351–1353. [Google Scholar] [CrossRef]
- Rieger, M.J.; Stolz, S.M.; Ludwig, S.; Benoit, T.M.; Bissig, M.; Widmer, C.C.; Schwotzer, R.; Müller, A.M.; Nair, G.; Hegemann, I.; et al. Daratumumab in rituximab-refractory autoimmune haemolytic anaemia. Br. J. Haematol. 2021, 194, 931–934. [Google Scholar] [CrossRef] [PubMed]
- McGlothlin, J.; Abeykoon, J.; Al-Hattab, E.; Ashrani, A.A.; Elliott, M.; Hook, C.C.; Pardanani, A.; Pruthi, R.; Sridharan, M.; Wolanskyj, A.; et al. Bortezomib and daratumumab in refractory autoimmune hemolytic anemia. Am. J. Hematol. 2023, 7. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Safety, Pharmacokinetics, and Efficacy of Subcutaneous Isatuximab in Adults with Warm Autoimmune Hemolytic Anemia (wAIHA). National Library of Medicine: Bethesda, MD, USA, 2023. [Google Scholar]
- McAlister, R.K.; Talbott, M.S.; Reddy, N.M. Durable responses in refractory autoimmune hemolytic anemia with alemtuzumab. J. Oncol Pharm. Pract. 2019, 25, 706–709. [Google Scholar] [CrossRef]
- Willis, F.; Marsh, J.C.; Bevan, D.H.; Killick, S.B.; Lucas, G.; Griffiths, R.; Ouwehand, W.; Hale, G.; Waldmann, H.; Gordon-Smith, E.C. The effect of treatment with Campath-1H in patients with autoimmune cytopenias. Br. J. Haematol. 2001, 114, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Rodon, P.; Breton, P.; Courouble, G. Treatment of pure red cell aplasia and autoimmune haemolytic anaemia in chronic lymphocytic leukaemia with Campath-1H. Eur. J. Haematol. 2003, 70, 319–321. [Google Scholar] [CrossRef]
- Cheung, W.W.; Hwang, G.Y.; Tse, E.; Kwong, Y.L. Alemtuzumab induced complete remission of autoimmune hemolytic anemia refractory to corticosteroids, splenectomy and rituximab. Haematologica 2006, 91, ECR13. [Google Scholar]
- Lundin, J.; Karlsson, C.; Celsing, F. Alemtuzumab therapy for severe autoimmune hemolysis in a patient with B cell chronic lymphocytic leukemia. Med. Oncol. 2006, 23, 137–139. [Google Scholar] [CrossRef]
- Karlsson, C.; Hansson, L.; Celsing, F.; Lundin, J. Treatment of severe refractory autoimmune hemolytic anemia in B-cell chronic lymphocytic leukemia with alemtuzumab (humanized CD52 monoclonal antibody). Leukemia 2007, 21, 511–514. [Google Scholar] [CrossRef]
- Royer, B.; Vaida, I.; Etienne, A.; Garidi, R.; Damaj, G.; Marolleau, J.P. Treatment of severe autoimmune hemolytic anemia in B-cell chronic lymphocytic leukemia with alemtuzumab. Leukemia 2007, 21, 1841–1842. [Google Scholar] [CrossRef][Green Version]
- Laurenti, L.; Tarnani, M.; Efremov, D.G.; Chiusolo, P.; De Padua, L.; Sica, S.; Leone, G. Efficacy and safety of low-dose alemtuzumab as treatment of autoimmune hemolytic anemia in pretreated B-cell chronic lymphocytic leukemia. Leukemia 2007, 21, 1819–1821. [Google Scholar] [CrossRef]
- Barcellini, W.; Murakhovskaya, I.; Terriou, L.; Pane, F.; Patriarca, A.; Butler, K.; Moran, S.; Wei, S.; Jäger, U. Long-term efficacy and safety results from an ongoing open-label phase 2 study of parsaclisib for the treatment of autoimmune hemolytic anemia (Aiha). HemaSphere 2022, 6, 186–187. [Google Scholar] [CrossRef]
- Lacerda, M.P.; Guedes, N.R.; Yamakawa, P.E.; Pereira, A.D.; Fonseca, A.R.B.M.D.; Chauffaille, M.L.L.F.; Goncalves, M.V.; Yamamoto, M.; Rodrigues, C.A. Treatment of refractory autoimmune hemolytic anemia with venetoclax in relapsed chronic lymphocytic leukemia with del(17p). Ann. Hematol. 2017, 96, 1577–1578. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.J.; Maldonado, E.; Danilov, A.V. Refractory Autoimmune Cytopenias Treated with Venetoclax. Hemasphere 2019, 4, e202. [Google Scholar] [CrossRef]
- Grossi, F.; Shum, M.K.; Gertz, M.A.; Roman, E. Inhibition of C3 with APL-2 Controls Hemolysis and Increases Hemoglobin Levels in Subjects with Autoimmune Hemolytic Anemia (AIHA). Blood 2018, 132, 3623. [Google Scholar] [CrossRef]
- de Boer, E.C.W.; Jalink, M.; Delvasto-Nuñez, L.; Meulenbroek, E.M.; Baas, I.; Janssen, S.R.; Folman, C.C.; Gelderman, K.A.; Wouters, D.; Engel, M.D.; et al. C1-inhibitor treatment in patients with severe complement-mediated autoimmune hemolytic anemia. Blood Adv. 2023, 7, 3128–3139. [Google Scholar] [CrossRef] [PubMed]
- Allgood, J.W.; Chaplin, H., Jr. Idiopathic acquired autoimmune hemolytic anemia. Areview of forty-seven cases treated from 1955 through 1965. Am. J. Med. 1967, 43, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Coon, W.W. Splenectomy in the treatment of hemolytic anemia. Arch. Surg. 1985, 120, 625–628. [Google Scholar] [CrossRef]
- Akpek, G.; McAneny, D.; Weintraub, L. Comparative response to splenectomy in Coombs-positive autoimmune hemolytic anemia with or without associated disease. Am. J. Hematol. 1999, 61, 98–102. [Google Scholar] [CrossRef]
- Weinmann, M.; Becker, G.; Einsele, H.; Bamberg, M. Clinical indications and biological mechanisms of splenic irradiation in autoimmune diseases. Strahlenther. Onkol 2001, 177, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Milosavljevic, V.; Tadic, B.; Grubor, N.; Eric, D.; Reljic, M.; Matic, S. Analysis of the surgical treatment of the patients operated on by using laparoscopic and classic splenectomy due to benign disorders of the spleen. Turk. J. Surg. 2019, 35, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Walsh, R.M.; McHam, S.; Brody, F.; Kalaycio, M. Laparoscopic splenectomy for autoimmune hemolytic anemia in patients with chronic lymphocytic leukemia: A case series and review of the literature. Am. J. Hematol. 2004, 75, 134–138. [Google Scholar] [CrossRef]
- Rosen, M.; Brody, F.; Walsh, R.M.; Tarnoff, M.; Malm, J.; Ponsky, J. Outcome of laparoscopic splenectomy based on hematologic indication. Surg. Endosc. 2002, 16, 272–279. [Google Scholar] [CrossRef]
- Balagué, C.; Targarona, E.M.; Cerdán, G.; Novell, J.; Montero, O.; Bendahan, G.; García, A.; Pey, A.; Vela, S.; Diaz, M.; et al. Long-term outcome after laparoscopic splenectomy related to hematologic diagnosis. Surg. Endosc. 2004, 18, 1283–1287. [Google Scholar] [CrossRef]
- Patel, N.Y.; Chilsen, A.M.; Mathiason, M.A.; Kallies, K.J.; Bottner, W.A. Outcomes and complications after splenectomy for hematologic disorders. Am. J. Surg. 2012, 204, 1014–1019, discussion 1019–1020. [Google Scholar] [CrossRef]
- Giudice, V.; Rosamilio, R.; Ferrara, I.; Seneca, E.; Serio, B.; Selleri, C. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med. 2016, 11, 374–380. [Google Scholar] [CrossRef]
- Lechner, K.; Jäger, U. How I treat autoimmune hemolytic anemias in adults. Blood 2010, 116, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Guédon, A.; Ribeil, J.A.; Suarez, F.; Tamburini, J.; Gaujoux, S. Indications and outcome of splenectomy in hematologic disease. J. Visc. Surg. 2017, 154, 421–429. [Google Scholar] [CrossRef]
- Sys, J.; Provan, D.; Schauwvlieghe, A.; Vanderschueren, S.; Dierickx, D. The role of splenectomy in autoimmune hematological disorders: Outdated or still worth considering? Blood Rev. 2017, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Maskal, S.; Al Marzooqi, R.; Fafaj, A.; Zolin, S.; Naples, R.; Iyer, A.; Petro, C.; Krpata, D.; Prabhu, A.; Rosen, M.; et al. Clinical and surgical outcomes of splenectomy for autoimmune hemolytic anemia. Surg. Endosc. 2022, 36, 5863–5872. [Google Scholar] [CrossRef]
- Barron, N.; Arenas-Osuna, J.; Medina, G.; Cruz-Dominguez, M.P.; González-Romero, F.; Velásques-García, J.A.; Ayala-López, E.A.; Jara, L.J. Splenectomy in systemic lupus erythematosus and autoimmune hematologic disease: A comparative analysis. Clin. Rheumatol. 2018, 37, 943–948. [Google Scholar] [CrossRef]
- Ogbue, O.; Bahaj, W.; Kewan, T.; Ahmed, R.; Ullah, F.; Visconte, V.; Maskal, S.; Gurnari, C.; Rosenblatt, S.; Maciejewski, J.P. Splenectomy for Immune Cytopenias: Treatment Outcomes and Predictors of Response. Blood 2022, 140, 1226–1227. [Google Scholar] [CrossRef]
- Sulpizio, E.D.; Raghunathan, V.; Shatzel, J.J.; Zilberman-Rudenko, J.; Worrest, T.; Sheppard, B.C.; DeLoughery, T.G. Long-term remission rates after splenectomy in adults with Evans syndrome compared to immune thrombocytopenia: A single-center retrospective study. Eur. J. Haematol. 2020, 104, 55–58. [Google Scholar] [CrossRef]
- Giannotta, J.A.; Fattizzo, B.; Cavallaro, F.; Barcellini, W. Infectious Complications in Autoimmune Hemolytic Anemia. J. Clin. Med. 2021, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.; Brunson, A.; Keegan, T.H.M.; Wun, T. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with autoimmune hemolytic anemia. Blood Cells Mol. Dis. 2020, 81, 102388. [Google Scholar] [CrossRef]
- Leone, G.; Pizzigallo, E. Bacterial Infections Following Splenectomy for Malignant and Nonmalignant Hematologic Diseases. Mediterr. J. Hematol. Infect. Dis. 2015, 7, e2015057. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.W.; Crowell, K.; Cravens, D. Clinical inquiries. Which vaccinations are indicated after splenectomy? J. Fam. Pract. 2006, 55, 711–712. [Google Scholar] [PubMed]
- Lee, G.; Malpica Castillo, L.E.; Walter, J.E. Infection Risk, Immunization Recommendations, and Antimicrobial Prophylaxis Needs When Treating Non-Malignant Hematologic Disorders—Wash Your Hands and What Else? Education Program. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Lecouffe-Desprets, M.; Néel, A.; Graveleau, J.; Leux, C.; Perrin, F.; Visomblain, B.; Artifoni, M.; Masseau, A.; Connault, J.; Pottier, P.; et al. Venous thromboembolism related to warm autoimmune hemolytic anemia: A case-control study. Autoimmun. Rev. 2015, 14, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.; Bret, P.M.; Atri, M.; Hreno, A.; Casola, G.; Gianfelice, D. Splenic vein thrombosis after splenectomy: Frequency and role of imaging. Radiology 1994, 190, 65–68. [Google Scholar] [CrossRef]
- Hassn, A.M.; Al-Fallouji, M.A.; Ouf, T.I.; Saad, R. Portal vein thrombosis following splenectomy. Br. J. Surg. 2000, 87, 362–373. [Google Scholar] [CrossRef]
- Okamoto, S.; Urade, T.; Yakushijin, K.; Kido, M.; Kuramitsu, K.; Komatsu, S.; Gon, H.; Yamashita, H.; Shirakawa, S.; Tsugawa, D.; et al. Successful Management of Refractory Autoimmune Hemolytic Anemia with Cold Agglutinin Disease with Splenectomy: A Case Report with Review of Literature. Kobe J. Med. Sci. 2023, 68, E30–E34. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| AIHA Subtype | Autoantibodies | Hemolysis | ||||
|---|---|---|---|---|---|---|
| Antibody Specificity | Thermal Amplitude | Specificity | DAT | |||
| wAIHA | IgG (+ possible C fixation) | 37 °C (0–40) 98.6 °F (32–104) | Rh system glycophorins A–D | IgG or IgG + C3d | Extravascular (mainly spleen) | |
| cAIHA | CAD | IgM, rare IgG (common C fixation) | 4 °C (4–34) 39.2 °F (39.2–93.2) | I/i system, rarely Pr or IH antigen | C3d CA titer ≥64 | Extravascular (liver) Intravascular |
| PCH | IgG, rare IgM (common C fixation) | react at 4 °C (39.2 °F) hemolyze at 37 °C (98.6 °F) | P antigen | C3d (Donath–Landsteiner test) | Intravascular | |
| Mixed AIHA | warm IgG and cold IgM | 4 °C (39.2 °F) and 37 °C (98.6 °F) | IgG + high-titer cold IgM | Intra- and extravascular | ||
| Drug/Mechanism of Action | Dose Schedule | Number of Patients | Response | Time to Response | Duration of Responses | Reference |
|---|---|---|---|---|---|---|
| Primary wAIHA | ||||||
| Fostamatinib/ syk inhibitor | 100–150 p.o. mg bid | 19 | 7/19 | 2–4 weeks | 24 weeks | Kuter et al. 2022 [62] |
| Parsaclisib/ PI3Kδ inhibitor | 1 mg p.o. qd 2.5 mg p.o. qd | 16 | 75% (PR 25%) | 2 weeks | 12 weeks | Barcellini et al. 2022 [90] |
| Bortezomib/Proteasome inhibitor | 1.3 mg/s.c. qw for 4 weeks + Rituximab | 1 | 1/1 | NA | 9 months | Ames et al. 2010 [75] |
| 1.3 mg/s.c. biw for 2 weeks + Rituximab | 4 | 4/4 | 1–3 weeks | 3–5 months | Chen et al. 2020 [76] | |
| 2.7 mg every 72 h total of four doses | 1 | 1/1 | 1 weeks | 7 months | Chineke et al. 2019 [77] | |
| Daratumumab/ antiDC38 mAb | 16 mg/kg qw for 6 weeks | 1 | 1/1 | NA | 5 months | Rieger et al. 2021 [79] |
| 16 mg/kg qw for 4 weeks | 1 | 1/1 | 12 weeks | 2 months | Jain et al. 2020 [78] | |
| 16 mg/kg qw for 2 months | 1 | 1/1 | 4 weeks | NA | McGlothlin et al. 2022 [80] | |
| Sirolimus/ mTOR inhibitor | 1–3 mg/d 6–18 months | 14 | 12/14 (4 PR) | 12–24 weeks | 16 months | Li et al. 2020 [69] |
| Secondary wAIHA | ||||||
| Alemtuzumab/ antiCD52 mAb wAIHA+CLL | 10 mg qd for 1.5 weeks | 2 | 2/2 (1 PR) | 8 weeks | 16 months | Willis et al. 2001 [83] |
| 30 mg d5, then 30 mg tiw for 3 weeks | 1 | 1/1 | 3 weeks | 10 months | Rondon et al. 2003 [84] | |
| 3 mg d1, 10 mg d3, and 30 mg tiw for 8 weeks | 1 | 1/1 | NA | 16 months | Cheung et al. 2006 [85] | |
| 10 mg s.c. d1, then 30 mg tiw for 8 weeks | 1 | 1/1 | 8 weeks | 15 months | Lundin et al. 2007 [86] | |
| 30 mg s.c. tiw 3–12 w for 8 weeks | 5 | 5/5 | 5 weeks (4–7) | 15 months | Karlsson et al. 2007 [87] | |
| 30 mg tiw: 8 weeks i.v.—episode 1 11 weeks s.c.—episode 2 | 1 | 1/1 | eeks—episode 1 NA—episode 2 | 17 months (episode 1) 3 months (episode 2) | Royer et al. 2007 [88] | |
| 3 mg d1-d3, then 10 mg tiw for 10–13 weeks | 3 | 3/3 | 5–8 weeks | 9–26 months | Leurenti et al. 2007 [89] | |
| after dose escalation, 30 mg tiw 8–16 weeks | 3 | 3/3 | NA | 7–68 months | Mc Alister et al. 2019 [82] | |
| Idelalisib/ PI3Kδ inhibitor | 150 mg p.o.bid + Rituximab | 12 | 11/12 (8 PR) | NA | median PFS not reached after 2 years | Quinquenel et al. 2019 [63] |
| Ibrutinib/ BTK inhibitor wAIHA+ CLL | 420 mg p.o. qd +/− Rituximab | 16 | 15/16 (8 PR) | NA | median PFS 19 months | Quinquenel et al. 2019 [63] |
| 420 mg p.o. +/− Rituximab | 8 | 6/8 | NA | 17.6 months | Vitale et al. 2016 [64] | |
| 420 mg p.o. qd + Prednisone 12.5 mg p.o. qd for 4 weeks | 1 | 1/1 | 5 weeks | 12 months | Cavazzini et al. 2016 [65] | |
| 420 mg p.o. + oral corticosteroids 6–17 weeks | 5 | 5/5 | 6–10 weeks | 15–150 weeks | Garcia-Horton A et al. 2016 [66] | |
| 420 mg + Prednisone ≤ 20 mg qd * | 21 | NA | NA | 17.5 months | Montilo et al. 2017 [67] | |
| Ibrutinib/ BTK inhibitor wAIHA+ MCL | 560 mg p.o. qd | 1 | 1/1 | 7 weeks | 6 months | Galinier et al. 2017 [68] |
| Venetoclax/ Bcl-2 inhibitor wAIHA+CLL | after standard ramp up, 400 mg p.o. qd | 1 | 1/1 | 3 months | 10 months | Lacerda et al. 2017 [91] |
| after standard ramp up, 400 mg p.o. qd | 1 | 1/1 | 4 weeks | 6 months | Gordon et al. 2019 [92] | |
| after standard ramp up, 400 mg p.o. qd + Rituximab | 2 | 2/2 | 3 months | 13 mo and 29 months | Galindo-Navarro et al. 2023 [73] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cvetković, Z.; Pantić, N.; Cvetković, M.; Virijević, M.; Sabljić, N.; Marinković, G.; Milosavljević, V.; Pravdić, Z.; Suvajdžić-Vuković, N.; Mitrović, M. The Role of the Spleen and the Place of Splenectomy in Autoimmune Hemolytic Anemia—A Review of Current Knowledge. Diagnostics 2023, 13, 2891. https://doi.org/10.3390/diagnostics13182891
Cvetković Z, Pantić N, Cvetković M, Virijević M, Sabljić N, Marinković G, Milosavljević V, Pravdić Z, Suvajdžić-Vuković N, Mitrović M. The Role of the Spleen and the Place of Splenectomy in Autoimmune Hemolytic Anemia—A Review of Current Knowledge. Diagnostics. 2023; 13(18):2891. https://doi.org/10.3390/diagnostics13182891
Chicago/Turabian StyleCvetković, Zorica, Nikola Pantić, Mirjana Cvetković, Marijana Virijević, Nikica Sabljić, Gligorije Marinković, Vladimir Milosavljević, Zlatko Pravdić, Nada Suvajdžić-Vuković, and Mirjana Mitrović. 2023. "The Role of the Spleen and the Place of Splenectomy in Autoimmune Hemolytic Anemia—A Review of Current Knowledge" Diagnostics 13, no. 18: 2891. https://doi.org/10.3390/diagnostics13182891
APA StyleCvetković, Z., Pantić, N., Cvetković, M., Virijević, M., Sabljić, N., Marinković, G., Milosavljević, V., Pravdić, Z., Suvajdžić-Vuković, N., & Mitrović, M. (2023). The Role of the Spleen and the Place of Splenectomy in Autoimmune Hemolytic Anemia—A Review of Current Knowledge. Diagnostics, 13(18), 2891. https://doi.org/10.3390/diagnostics13182891