
A Computational Approach to Identification of Candidate Biomarkers in High-Dimensional Molecular Data

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Feature Selection Algorithm
2.2. Data Acquisition
2.3. Data Preprocessing
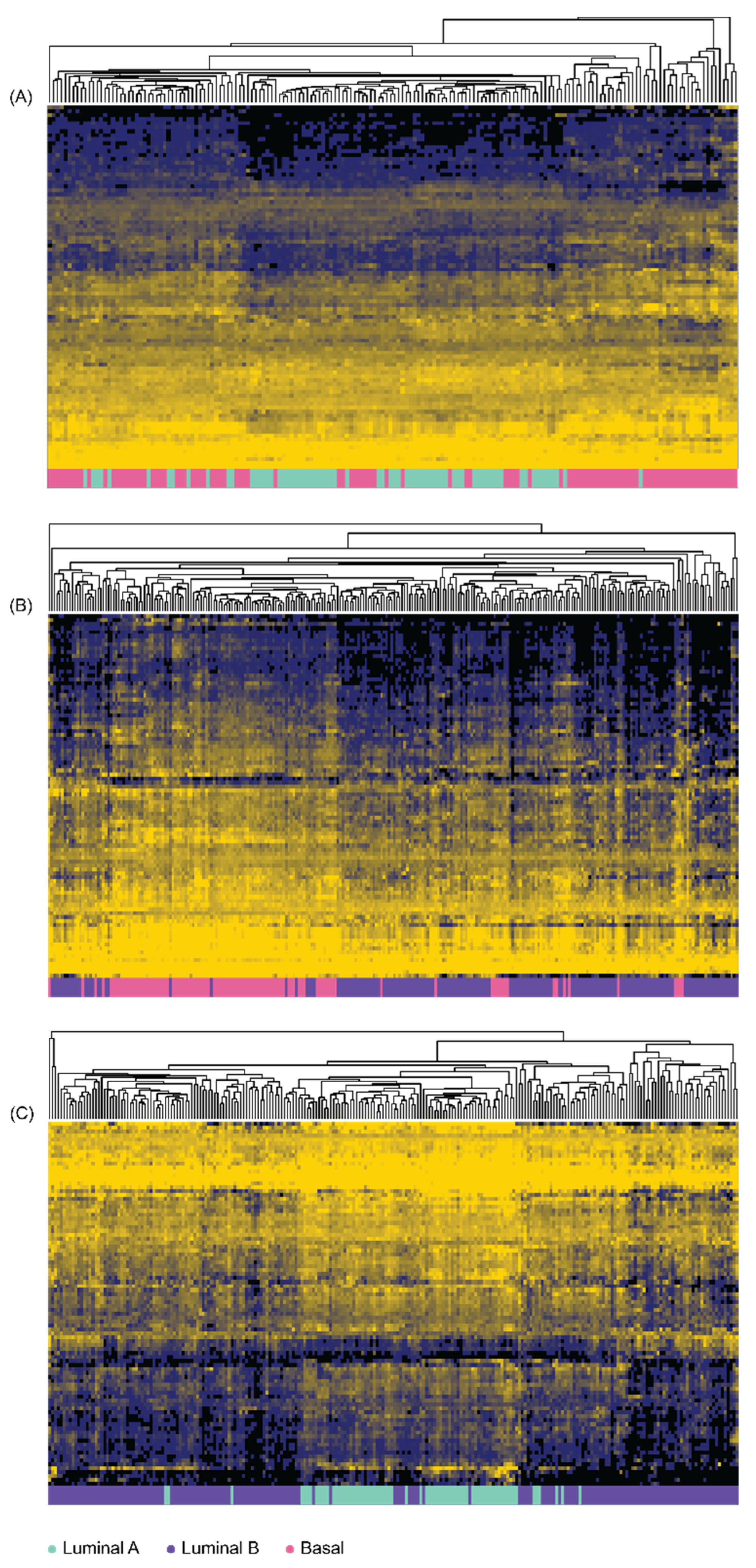
2.4. Prostate Cancer Stratification into Molecular Subtypes with Pam50
2.5. Immune Profile Compilation
2.6. Feature Selection of Immune-Function Genes in Prostate Adenocarcinoma
2.7. Differential Gene Expression of Immune-Function Genes in Prostate Adenocarcinoma
2.8. Construction of an Immune-Function Gene Classifier
2.9. Clinical Information Analyses
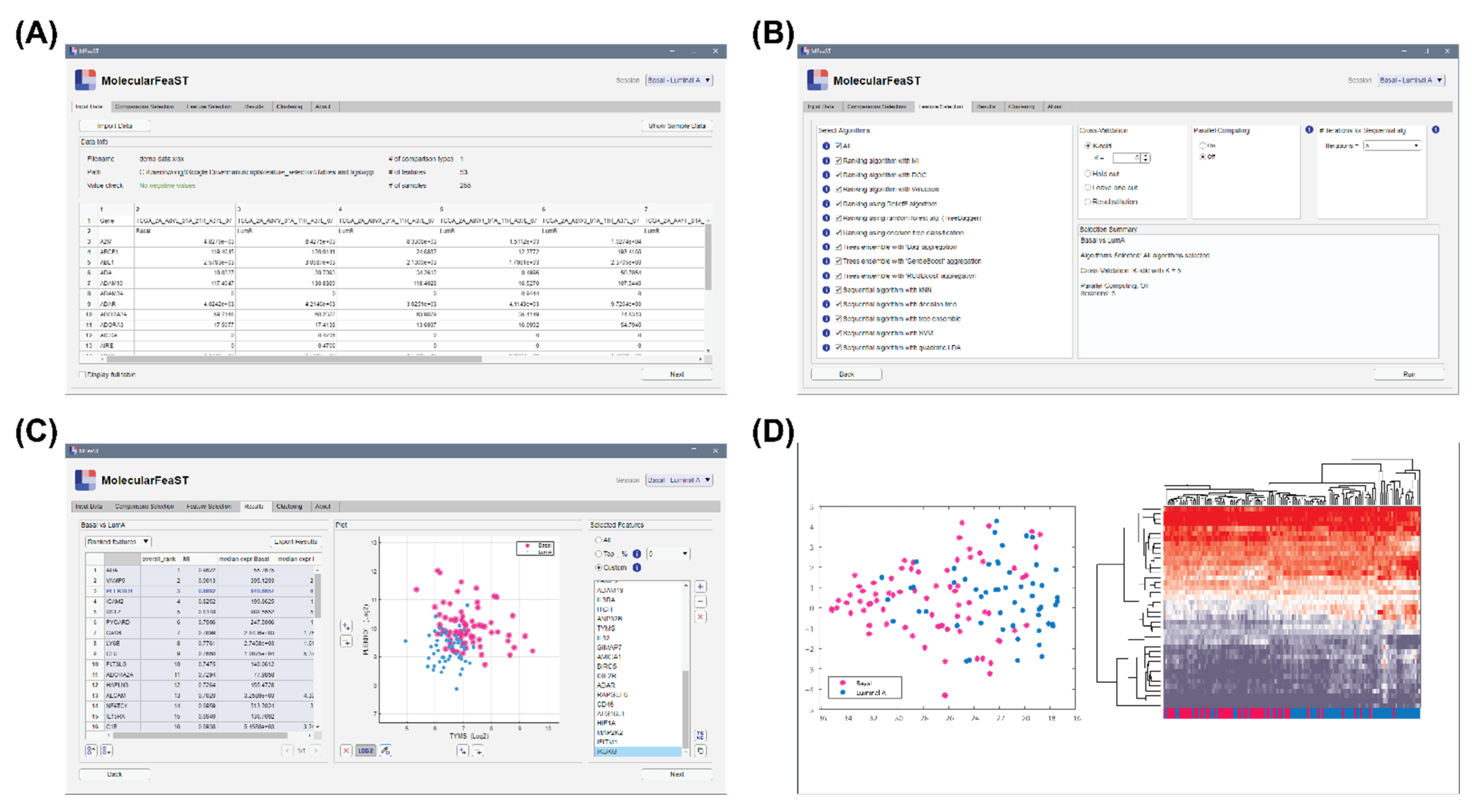
2.10. Graphical User Interface for the Feature Selection Algorithm
3. Results
3.1. Clinical Information Analyses
3.2. Feature Selection of Immune-Function Genes in Prostate Adenocarcinoma
3.3. Differential Gene Expression of Immune-Function Genes in Prostate Adenocarcinoma
3.4. Immune-Function Gene Classifier
3.5. MFeaST Graphical User Interface
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finotello, F.; Di Camillo, B. Measuring differential gene expression with RNA-seq: Challenges and strategies for data analysis. Brief. Funct. Genom. 2015, 14, 130–142. [Google Scholar] [CrossRef]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef]
- Sullivan, G.M.; Feinn, R. Using Effect Size-or Why the P Value Is Not Enough. J. Grad. Med. Educ. 2012, 4, 279–282. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Ellis, P.D. The Essential Guide to Effect Sizes: Statistical Power, Meta-Analysis, and the Interpretation of Research Results; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Hira, Z.M.; Gillies, D.F. A Review of Feature Selection and Feature Extraction Methods Applied on Microarray Data. Adv. Bioinform. 2015, 2015, 198363. [Google Scholar] [CrossRef]
- Duda, R.O.; Hart, P.E.; Stork, D.G. Pattern Classification, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Ao, S.-I. Data Mining and Applications in Genomics, 1st ed.; Springer: Heidelberg, Germany, 2008. [Google Scholar]
- Raudys, S.; Pikelis, V. On dimensionality, sample size, classification error, and complexity of classification algorithm in pattern recognition. IEEE Trans. Pattern. Anal. Mach. Intell. 1980, 2, 242–252. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef]
- Lazar, C.; Taminau, J.; Meganck, S.; Steenhoff, D.; Coletta, A.; Molter, C.; de Schaetzen, V.; Duque, R.; Bersini, H.; Nowe, A. A survey on filter techniques for feature selection in gene expression microarray analysis. IEEE ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1106–1119. [Google Scholar] [CrossRef]
- Remeseiro, B.; Bolon-Canedo, V. A review of feature selection methods in medical applications. Comput. Biol. Med. 2019, 112, 103375. [Google Scholar] [CrossRef]
- Tadist, K.; Najah, S.; Nikolov, N.S.; Mrabti, F.; Zahi, A. Feature selection methods and genomic big data: A systematic review. J. Big Data 2019, 6, 79. [Google Scholar] [CrossRef]
- Park, S.; Shin, B.; Sang Shim, W.; Choi, Y.; Kang, K.; Kang, K. Wx: A neural network-based feature selection algorithm for transcriptomic data. Sci. Rep. 2019, 9, 10500. [Google Scholar] [CrossRef]
- Han, H. A novel feature selection for RNA-seq analysis. Comput. Biol. Chem. 2017, 71, 245–257. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Abeel, T.; Helleputte, T.; Van de Peer, Y.; Dupont, P.; Saeys, Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 2010, 26, 392–398. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, X.; Xu, J.; Quan, X.; Wu, M.; Zhang, H. Ensemble Consensus-Guided Unsupervised Feature Selection to Identify Huntington’s Disease-Associated Genes. Genes 2018, 9, 350. [Google Scholar] [CrossRef]
- Moon, M.; Nakai, K. Stable feature selection based on the ensemble L 1-norm support vector machine for biomarker discovery. BMC Genom. 2016, 17 (Suppl. S13), 1026. [Google Scholar] [CrossRef]
- Bolón-Canedo, V.; Alonso-Betanzos, A. Ensembles for feature selection: A review and future trends. Inf. Fusion 2019, 52, 1–12. [Google Scholar] [CrossRef]
- Shahrjooihaghighi, A.; Frigui, H.; Zhang, X.; Wei, X.; Shi, B.; Trabelsi, A. An Ensemble Feature Selection Method for Biomarker Discovery. Proc. IEEE Int. Symp. Signal Proc. Inf. Tech. 2017, 2017, 416–421. [Google Scholar] [CrossRef]
- Plyushchenko, I.; Shakhmatov, D.; Bolotnik, T.; Baygildiev, T.; Nesterenko, P.N.; Rodin, I. An approach for feature selection with data modelling in LC-MS metabolomics. Anal. Methods 2020, 12, 3582–3591. [Google Scholar] [CrossRef]
- Cormen, T.H.; Leiserson, C.E.; Rivest, R.L.; Stein, C. 16 Greedy Algorithms. In Introduction to Algorithms; MIT Press: Cambridge, MA, USA, 2001; p. 370. [Google Scholar]
- Ren, R.; Tyryshkin, K.; Graham, C.H.; Koti, M.; Siemens, D.R. Comprehensive immune transcriptomic analysis in bladder cancer reveals subtype specific immune gene expression patterns of prognostic relevance. Oncotarget 2017, 8, 70982–71001. [Google Scholar] [CrossRef]
- Symons, L.K.; Miller, J.E.; Tyryshkin, K.; Monsanto, S.P.; Marks, R.M.; Lingegowda, H.; Vanderbeck, K.; Childs, T.; Young, S.L.; Lessey, B.A.; et al. Neutrophil recruitment and function in endometriosis patients and a syngeneic murine model. FASEB J. 2020, 34, 1558–1575. [Google Scholar] [CrossRef]
- Hamade, A.; Li, D.; Tyryshkin, K.; Xu, M.; Conseil, G.; Yolmo, P.; Hamilton, J.; Chenard, S.; Robert Siemens, D.; Koti, M. Sex differences in the aging murine urinary bladder and influence on the tumor immune microenvironment of a carcinogen-induced model of bladder cancer. Biol. Sex Differ. 2022, 13, 19. [Google Scholar] [CrossRef]
- Kim, H.K.; Tyryshkin, K.; Elmi, N.; Dharsee, M.; Evans, K.R.; Good, J.; Javadi, M.; McCormack, S.; Vaccarino, A.L.; Zhang, X.; et al. Plasma microRNA expression levels and their targeted pathways in patients with major depressive disorder who are responsive to duloxetine treatment. J. Psychiatr. Res. 2019, 110, 38–44. [Google Scholar] [CrossRef]
- Kim, H.K.; Tyryshkin, K.; Elmi, N.; Feilotter, H.; Andreazza, A.C. Examining redox modulation pathways in the post-mortem frontal cortex in patients with bipolar disorder through data mining of microRNA expression datasets. J. Psychiatr. Res. 2018, 99, 39–49. [Google Scholar] [CrossRef]
- Panarelli, N.; Tyryshkin, K.; Wong, J.J.M.; Majewski, A.; Yang, X.; Scognamiglio, T.; Kim, M.K.; Bogardus, K.; Tuschl, T.; Chen, Y.T.; et al. Evaluating gastroenteropancreatic neuroendocrine tumors through microRNA sequencing. Endocr. Relat. Cancer 2019, 26, 47–57. [Google Scholar] [CrossRef]
- Turashvili, G.; Lightbody, E.D.; Tyryshkin, K.; SenGupta, S.K.; Elliott, B.E.; Madarnas, Y.; Ghaffari, A.; Day, A.; Nicol, C.J.B. Novel prognostic and predictive microRNA targets for triple-negative breast cancer. FASEB J. 2018, 32, 5937–5954. [Google Scholar] [CrossRef]
- Nanayakkara, J.; Tyryshkin, K.; Yang, X.; Wong, J.J.M.; Vanderbeck, K.; Ginter, P.S.; Scognamiglio, T.; Chen, Y.-T.; Panarelli, N.; Cheung, N.-K.; et al. Characterizing and classifying neuroendocrine neoplasms through microRNA sequencing and data mining. NAR Cancer 2020, 2, zcaa009. [Google Scholar] [CrossRef]
- Wong, J.J.M.; Ginter, P.S.; Tyryshkin, K.; Yang, X.; Nanayakkara, J.; Zhou, Z.; Tuschl, T.; Chen, Y.T.; Renwick, N. Classifying Lung Neuroendocrine Neoplasms through MicroRNA Sequence Data Mining. Cancers 2020, 12, 2653. [Google Scholar] [CrossRef]
- Tyryshkin, K.; Li, Y.D.; Good, D.; Shepherd, L.E.; Baetz, T.; Rauh, M.J.; LeBrun, D.P. Differential Expression of TCF3 Target Genes Defines Subclasses of Diffuse Large B-Cell Lymphoma with Striking Differences in Clinical Outcome Following R-CHOP Therapy. Blood 2016, 128, 3037. [Google Scholar] [CrossRef]
- Grenier-Pleau, I.; Tyryshkin, K.; Le, T.D.; Rudan, J.; Bonneil, E.; Thibault, P.; Zeng, K.; Lasser, C.; Mallinson, D.; Lamprou, D.; et al. Blood extracellular vesicles from healthy individuals regulate hematopoietic stem cells as humans age. Aging Cell 2020, 19, e13245. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Hoaglin, D.C.; Iglewicz, B. Fine-Tuning Some Resistant Rules for Outlier Labeling. J. Am. Stat. Assoc. 1987, 82, 1147–1149. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Das, R.; Chang, S.L.; Tomlins, S.A.; Chou, J.; Quigley, D.A.; Dang, H.X.; Barnard, T.J.; Mahal, B.A.; et al. Clinical and Genomic Implications of Luminal and Basal Subtypes Across Carcinomas. Clin. Cancer Res. 2019, 25, 2450–2457. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9. [Google Scholar]
- Liang, S.; Ma, A.; Yang, S.; Wang, Y.; Ma, Q. A Review of Matched-pairs Feature Selection Methods for Gene Expression Data Analysis. Comput. Struct. Biotechnol. J. 2018, 16, 88–97. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Feature Selection Type | Univariable | Multivariable |
|---|---|---|
| Filter Type | Mutual information score ROC criteria Wilcoxon criteria ReliefF analysis | |
| Wrapper Type | Support vector machine K-nearest neighbors Decision tree Quadratic discriminant analysis | |
| Embedded Type | Treebagger predictor importance | Decision tree with bagging Decision tree with gentle adaptive boosting Decision tree with random undersampling boosting |
| Basal | Luminal A | Luminal B | Total | χ2 | D.o.F | p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biochemical recurrence | n = 88 | n = 64 | n = 144 | n = 296 | |||||||||||
| Yes | 14 (16%) | 3 (5%) | 24 (17%) | 41 (14%) | 5.77 | 2 | 0.056 | ||||||||
| No | 74 (84%) | 61 (95%) | 120 (83%) | 255 (86%) | |||||||||||
| Radiation therapy | n = 61 | n = 36 | n = 99 | n = 296 | |||||||||||
| Yes | 8 (13%) | 6 (17%) | 14 (14%) | 28 (9%) | 0.24 | 2 | 0.888 | ||||||||
| No | 53 (87%) | 30 (83%) | 85 (86%) | 168 (57%) | |||||||||||
| Radiation follow up | n = 80 | n = 64 | n = 132 | n = 276 | |||||||||||
| Yes | 9 (11%) | 8 (13%) | 25 (19%) | 42 (15%) | 2.76 | 2 | 0.252 | ||||||||
| No | 71 (89%) | 56 (88%) | 107 (81%) | 234 (85%) | |||||||||||
| Histological type | n = 102 | n = 72 | n = 166 | n = 340 | |||||||||||
| Acinar | 101 (99%) | 72 (100%) | 158 (95%) | 331 (97%) | * 6.10 | 2 | 0.047 | ||||||||
| Other | 1 (1%) | 0 (0%) | 8 (5%) | 9 (3%) | |||||||||||
| Grade group | n = 99 | n = 71 | n = 164 | n = 334 | |||||||||||
| GG1 | 10 (10%) | 8 (11%) | 11 (7%) | 29 (9%) | 21.221 | 6 | 0.020 | ||||||||
| GG2 | 35 (35%) | 28 (39%) | 31 (19%) | 94 (28%) | |||||||||||
| GG3 | 17 (17%) | 14 (20%) | 30 (18%) | 61 (18%) | |||||||||||
| GG4 + GG5 | 37 (37%) | 21 (30%) | 92 (56%) | 150 (45%) | |||||||||||
| Pathological stage | n = 100 | n = 69 | n = 166 | n = 335 | |||||||||||
| pT2 | 41 (41%) | 35 (51%) | 50 (30%) | 126 (38%) | 9.515 | 2 | 0.009 | ||||||||
| pT3 + pT4 | 59 (59%) | 34 (49%) | 116 (70%) | 209 (62%) | |||||||||||
| Nodal involvement | n = 83 | n = 62 | n = 151 | n = 296 | |||||||||||
| pN0 | 73 (88%) | 52 (84%) | 114 (75%) | 239 (81%) | 5.84 | 2 | 0.054 | ||||||||
| pN1 | 10 (12%) | 10 (16%) | 37 (25%) | 57 (19%) | |||||||||||
| Basal | Luminal A | Luminal B | Total | ||||||||||||
| med | n | ran | med | n | ran | med | n | ran | med | n | ran | H | D.o.F | p | |
| Age at diagnosis | 62 | 102 | 41–77 | 63 | 72 | 46–75 | 62 | 166 | 46–78 | 62 | 340 | 41–78 | 0.17 | 2 | 0.918 |
| PSA | 0.1 | 90 | 0–37.36 | 0.1 | 68 | 0–13.95 | 0.1 | 143 | 0–39.80 | 0.1 | 301 | 0–39.80 | 1.93 | 2 | 0.381 |
| MFeaST | Differential Expression | All Features | |
|---|---|---|---|
| (A) Basal|Luminal A | |||
| Number of genes | 33 | 295 | 574 |
| Accuracy | 81.08 ± 11.71 | 78.82 ± 9.77 | 77.12 ± 8.96 |
| Precision | 79.59 ± 19.84 | 78.03 ± 17.63 | 73.95 ± 12.80 |
| Recall | 82.86 ± 16.22 | 77.68 ± 13.81 | 75.00 ± 14.31 |
| F1-score | 78.86 ± 11.35 | 75.58 ± 8.80 | 73.09 ± 9.15 |
| (B) Basal|Luminal B | |||
| Number of genes | 18 | 472 | 574 |
| Accuracy | 94.80 ± 2.58 | 92.91 ± 6.18 | 94.02 ± 6.61 |
| Precision | 96.56 ± 3.88 | 93.46 ± 7.36 | 93.53 ± 7.37 |
| Recall | 95.22 ± 3.75 | 95.74 ± 4.17 | 97.50 ± 4.37 |
| F1-score | 95.79 ± 2.07 | 94.42 ± 4.68 | 95.33 ± 5.03 |
| (C) Luminal A|Luminal B | |||
| Number of genes | 15 | 328 | 574 |
| Accuracy | 95.36 ± 3.69 | 91.59 ± 5.20 | 92.45 ± 6.45 |
| Precision | 96.65 ± 5.18 | 93.35 ± 5.84 | 94.50 ± 6.36 |
| Recall | 96.99 ± 3.18 | 95.22 ± 6.09 | 95.26 ± 6.08 |
| F1-score | 96.70 ± 2.56 | 94.05 ± 3.68 | 94.66 ± 4.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerolami, J.; Wong, J.J.M.; Zhang, R.; Chen, T.; Imtiaz, T.; Smith, M.; Jamaspishvili, T.; Koti, M.; Glasgow, J.I.; Mousavi, P.; et al. A Computational Approach to Identification of Candidate Biomarkers in High-Dimensional Molecular Data. Diagnostics 2022, 12, 1997. https://doi.org/10.3390/diagnostics12081997
Gerolami J, Wong JJM, Zhang R, Chen T, Imtiaz T, Smith M, Jamaspishvili T, Koti M, Glasgow JI, Mousavi P, et al. A Computational Approach to Identification of Candidate Biomarkers in High-Dimensional Molecular Data. Diagnostics. 2022; 12(8):1997. https://doi.org/10.3390/diagnostics12081997
Chicago/Turabian StyleGerolami, Justin, Justin Jong Mun Wong, Ricky Zhang, Tong Chen, Tashifa Imtiaz, Miranda Smith, Tamara Jamaspishvili, Madhuri Koti, Janice Irene Glasgow, Parvin Mousavi, and et al. 2022. "A Computational Approach to Identification of Candidate Biomarkers in High-Dimensional Molecular Data" Diagnostics 12, no. 8: 1997. https://doi.org/10.3390/diagnostics12081997
APA StyleGerolami, J., Wong, J. J. M., Zhang, R., Chen, T., Imtiaz, T., Smith, M., Jamaspishvili, T., Koti, M., Glasgow, J. I., Mousavi, P., Renwick, N., & Tyryshkin, K. (2022). A Computational Approach to Identification of Candidate Biomarkers in High-Dimensional Molecular Data. Diagnostics, 12(8), 1997. https://doi.org/10.3390/diagnostics12081997