1. Introduction
Multiple Sclerosis (MS) is a chronic inflammatory disease that attacks the central nervous system. MS is characterized by lesions in the brain and spinal cord, which cause various neurological symptoms, including, but not limited to blindness, double vision, muscle weakness, and changes in sensation and balance [
1]. Multiple sclerosis ranges from the relapsing form, where attacks are over long intervals, to the progressive form, where symptoms progressively get worse over time. It is classified into Clinically Isolated Syndrome (CIS), Relapsing–Remitting MS (RRMS), Primary Progressive MS (PPMS), and Secondary Progressive MS (SPMS) [
2]. To the present date, there is no known cure for MS, nor the causes of the disease. Some mention the cause to be the destruction by the immune system or the failure of cells that produce myelin to protect the nerves; others mention genetics or viral infection [
3].
Multiple sclerosis is known to be one of the most common auto-immune disorders. The reports about the global burden of disease [
4] revealed that, in 2015, about 2.3 million people were diagnosed as MS patients, with about 18,900 deaths, as opposed to 12,000 deaths from MS in 1990.
In [
5], a study involving patients with Pediatric-Onset MS (POMS) was carried out to evaluate changes in the prognosis of POMS over time with associated therapeutic changes. It was shown that the risk of persistent disability was reduced by 50% to 70% in recent diagnosis epochs. Furthermore, in [
6], Late-Onset Relapsing–Remitting MS (LORRMS) and Young-Onset Relapsing–Remitting MS (YORRMS) were studied, and it was shown that the male population with LORRMS reached severe disability faster than those with YORRMS. Moreover, [
7] studied more symptoms associated with MS. The study showed that severe disease course is associated with a higher risk of neuropathic pain. Furthermore, as shown in [
8], dysphagia is one of the frequent MS symptoms.
This work is motivated by the fact that patients have an increased risk of being poorly diagnosed, hence increasing the risk factors and deterioration due to attacks. In addition to the availability of data and the increased diagnostic performance achieved by machine learning, machine learning has recently shown promising results for many neurological disorders. Proposed methods have successfully distinguished healthy subjects from patients with high accuracy. However, none of the proposed diagnostic methods have achieved reliable levels of accuracy to identify the genes associated with the disease as a basis for diagnosis. Moreover, none of the work in the literature has addressed the class imbalance nature of most of the MS expression data samples. Furthermore, most of these studies rely on data that are rarely used in clinical routine. In addition to that, ensemble learning has proven to be an effective approach for improving disease diagnosis accuracy.
Many researchers addressed the problem of multiple sclerosis prediction from different perspectives. However, the literature still lacks efficient predictive models that benefit from all the available data. In this section, the most cited and recent work in multiple sclerosis is summarized. Furthermore, the section briefly covers the work related to the imbalance dataset problem in diseases. Some work in the literature addressed the association of certain genes with the disease. Random forests were used in [
9] to identify new genes associated with MS.
Weygandt et al. [
10] used SVM to classify Relapsing–Remitting MS (RRMS) patients. The highest accuracy reported on 44 patients and 26 healthy controls was 95% using brain lesions, given that lesions are much more frequent in MS patients than in healthy controls, and also, normal-appearing tissue can be affected by microstructural changes [
11].
Bendefeldt et al., in [
12], employed Support Vector Machines (SVMs) to perform binary classification in MS to classify patients with a short disease duration (less than 5 years) and a long disease duration (more than 10 years), low T2 lesion load (less than 1 mL) and high T2 lesion load (more than 10 mL), and benign MS (with an Expanded Disability Status Scale (EDSS) less than or equal to 3) and non-benign MS (EDSS more than 3). The accuracy reported was 85%, 83%, and 77%, respectively.
Chen et al. [
13] proposed the Voxelwise Displacement Classifier (VDC); a classifier based on Fisher’s linear discriminant analysis, SVM, Random Forest(RF)and Adaboost when using displacement fields as features. The study was tested on 29 Relapse–Remitting MS (RRMS), 8 Secondary Progressive MS (SPMS), 4 CIS, and 1 Primary Progressive MS (PPMS) patients and 36 healthy controls. The proposed VD classifier consistently outperformed other methods, reaching up to 100% accuracy.
Other methods used gene expression data for diagnosing multiple sclerosis. The study [
14] proposed a classification model for gene selection using gene expression data, The method proposed was applied on a total of 44 samples, 26 multiple sclerosis patients and 18 individuals with other neurological diseases (control). An accuracy of 86% was achieved using an analytical framework integrating feature ranking algorithms and a support vector machine model for selecting genes associated with multiple sclerosis.
Sweeney et al. [
15] compared a supervised machine learning techniques using different feature vectors extracted from MRI images to predict MS. The research concluded that the choice of the feature vector has a higher impact on the predictive performance than the machine learning algorithm used.
A method for the diagnosis of multiple sclerosis using combined clinical data with lesion loads and magnetic resonance metabolic features was presented in [
16]. Three classifiers were used in the study, Linear Discriminant Analysis (LDA), Support Vector Machines (SVMs), and Random Forest (RF). The results reported in the paper suggest that metabolic features obtain good results to discriminate between relapsing–remitting and primary progressive forms, while lesion loads are better at discriminating between relapsing–remitting and secondary progressive forms. Therefore, combining clinical data with magnetic resonance lesion loads and metabolic features can improve the discrimination between relapsing–remitting and progressive forms.
Margineau et al. [
17] used four binary classifiers to classify multiple sclerosis courses using features extracted from Magnetic Resonance Spectroscopic Imaging (MRSI) combined with brain tissue segmentations of gray matter, white matter, and lesions. Values of the area under the curve ranged between 68% and 95%, with the highest percentage recorded for Support Vector Machines with a Gaussian kernel (SVM-rbf) applied on MRSI features combined with brain tissue segmentation features. According to their comparison, their work concluded that combining metabolic ratios with brain tissue segmentation percentages obtained high classification results for Clinically Isolated Syndrome (CIS), Relapsing–Remitting (RR), and Primary Progressive (PP) patients. The best results were obtained with SVM-rbf, and therefore, building complex architectures of convolutional neural networks does not add any improvement over classical machine learning methods.
Ostemeyer et al. [
18] used statistical learning to diagnose immune diseases. Their method was a repertoire-based statistical classifier for diagnosing Relapsing–Remitting Multiple Sclerosis (RRMS) with an accuracy up to 87%. Moreover, this method points to a diagnostic biochemical motif in the antibodies of RRMS patients. Zhao et al. [
19] used SVMs and compared them to logistic regression on 1693 CLIMB patients using demographic, clinical, and MRI data. The study showed that SVMs improved predictions and outperformed linear regression in most cases.
In [
20], machine learning techniques were employed on microarray datasets to identify defective pathways related to MS. The analysis resulted in a list of highly discriminatory genes, where the most discriminatory genes were related to the production of Hemoglobin. The analysis also revealed coincidences of MS with some viruses, such as Epstein–Barr virus, Influenza A, Toxoplasmosis, Tuberculosis, and Staphylococcus Aureus infections.
MS could also be diagnosed with some biomarkers as shown in the work in [
21]. The biomarkers suggested in [
21] are the plasma levels of Tumor Necrosis Factor (TNF)-
, soluble TNF Receptor (sTNFR) 1, sTNFR2, adiponectin, hydroperoxides, Advanced Oxidation Protein Products (AOPPs), nitric oxide metabolites, total plasma antioxidant capacity using the Total Radical-trapping Antioxidant Parameter (TRAP), Sulfhydryl (SH) groups, and serum levels of zinc. Support vector machines were used on 174 MS patients and 182 controls and achieved a training accuracy of 92.9% and a validation accuracy of 90.6%. The results showed that MS is characterized by lower levels of zinc, adiponectin, TRAP, and SH groups and higher levels of AOPPs.
In 2020, Zhao et al. [
22] extended the work performed in 2017 in [
19] and used 724 patients from the Comprehensive Longitudinal Investigation in MS at Brigham and Women’s Hospital (CLIMB study) and 400 patients from the EPIC dataset, University of California, San Francisco, to continue the study on MS prediction. This paper used SVM, logistic regression, and random forest, in addition to the ensemble learning approaches XGBoost, LightGBM, and Meta-learner L. The research concluded that ensemble methods give higher predictive performance of MS disease.
In [
23], support vector machines were used to diagnose MS using the plasma levels of selenium, vitamin B12, and vitamin D3. The study used 99 MS patients and 81 healthy controls. The supervised machine learning methods used were the support vector machine algorithm, decision tree, and K-nearest-neighbor. The highest accuracy, 98.89%, was achieved using support vector machines.
Shang et al. [
24] used gene expression data to uncover genes associated with MS. the authors performed bioinformatics analysis to identify differentially expressed genes and also explored the potential SNPs associated with MS. The study provided identified genes, SNPs, biological processes, and cellular pathways associated with MS.
Another important aspect considered in this paper is imbalanced data. A dataset is considered to be imbalanced when the number of samples representing one class is significantly fewer than the other classes. The class with the fewest number of samples is called the minority class, and the others are the majority classes. Handling the class imbalance data problem is of great interest, as shows in many real-world problems, especially medical diagnosis and disease prediction. Training classifiers on these datasets makes the classifiers more biased towards the majority classes, as the rules that predict the majority class samples are positively weighted, whereas the rules that predict the minority class are usually treated as noise or ignored. This leads to the conclusion that the minority class samples are prone to misclassification [
25]. Class-imbalance-aware methods either modify the standard classifiers used or incorporate a data-driven approach in the training process to deal with the different class sample sizes. Under-sampling and over-sampling are the data-driven techniques most commonly used for handling class imbalance datasets. Recently, much research has been presented to handle this problem. For instance, Garcia et. al. [
26] presented a data-level ensemble approach for class imbalance data based on GACE meta-heuristics and feature space adaptive partitioning. From the results, this system was able to reduce the time complexity and improve the imbalanced classification accuracy. An approach presented by K. Pasupa et. al. [
27] modified the stander classification process for the Convolutions Neural Network (CNN). A focal loss function for a classification task was employed to handle the data imbalance problem in a CNN deep learning model. Comparing the focal loss function with the cross-entropy function proved that the focal loss function can enable the deep learning model to be less biased towards the class with majority samples.
One of the flaws of these algorithm-based methods is that they require specific modifications to the classification algorithm, which makes it lack generality and a systematic approach for evaluating the performance of different classifiers. Classification methods are developed to predict the class of the future samples based on the assumption that the training samples are good examples of the future samples, where matching the frequency of classes in training data would warrant a realistic performance. Moreover, data-driven down-sampling methods lead to missing some of the majority class samples, which could have significant information for the classifier.
It is concluded from the background and related work discussed in the Introduction above that the diagnosis of MS is an extensible process with variable possible types of data that can be used, as well as several techniques. In the proposed model, we relied on gene expression data to diagnose MS based on the fact that gene expression identifies a unique group of genes in a cell that occurs as a result of an altered or unaltered biological process or pathogenic medical condition. As most of the expression profile datasets available for MS are imbalanced, our proposed model adapts an over-sampling technique to handle class imbalance expression data.
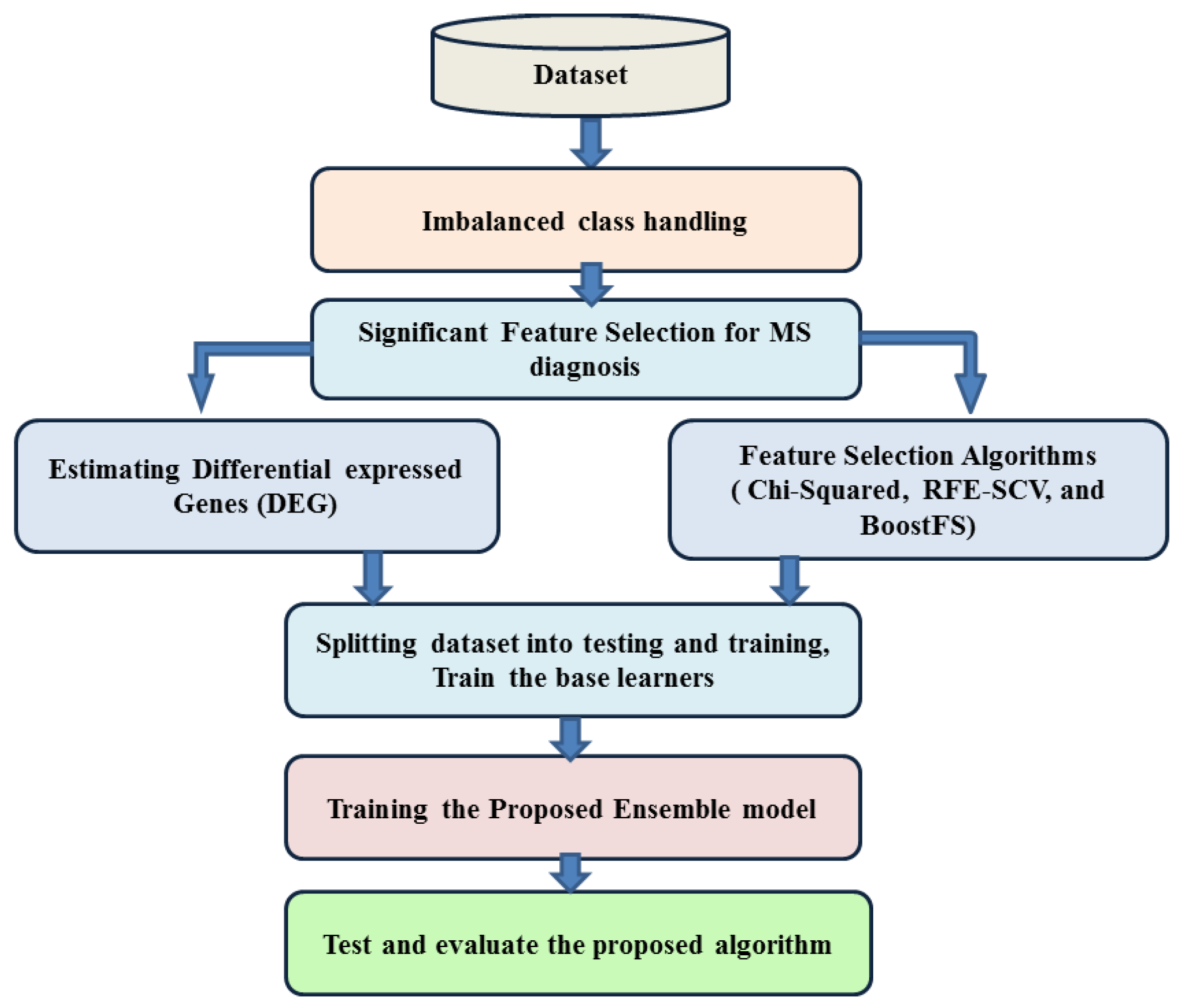
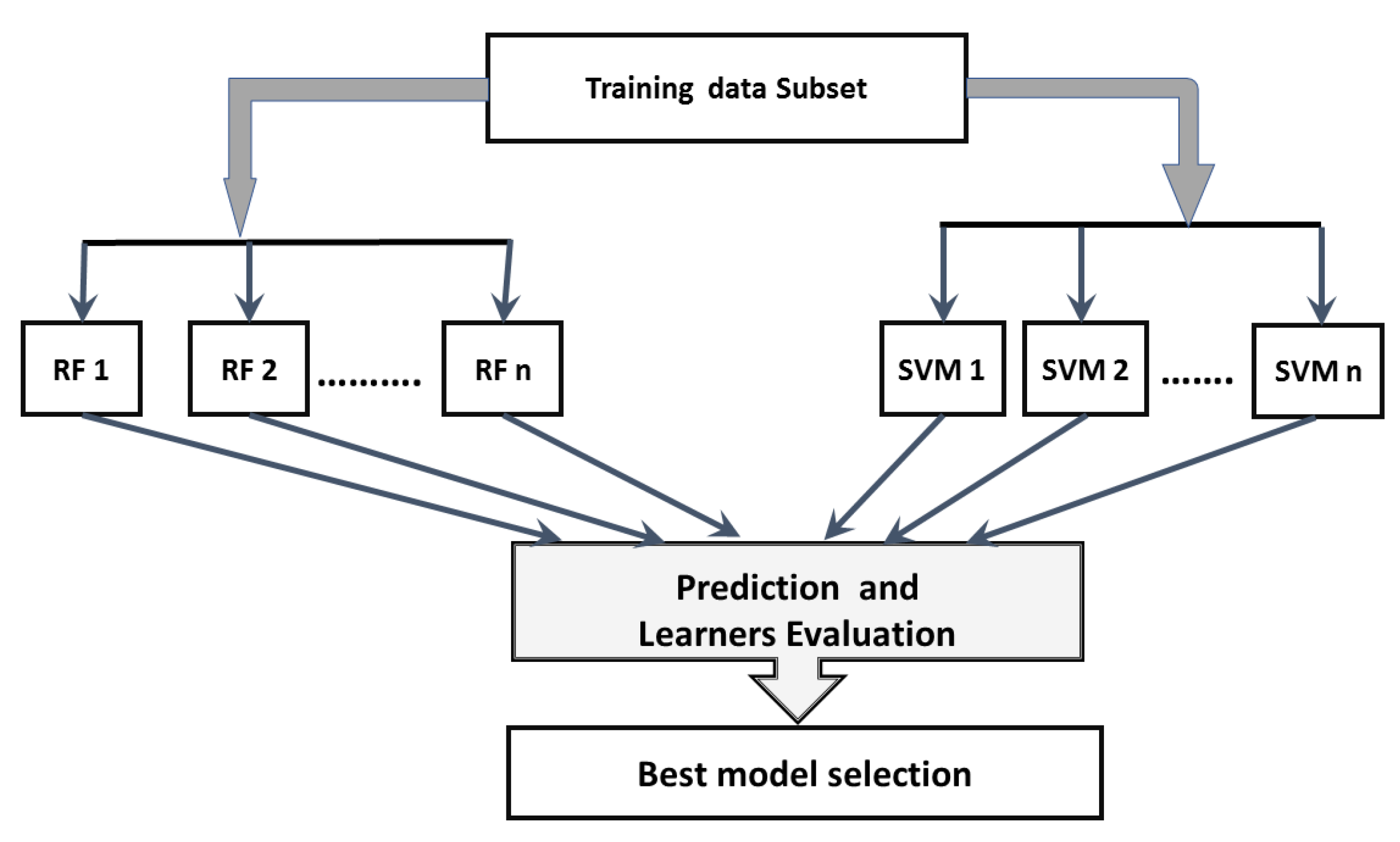
In this paper, a hierarchical ensemble approach that employs voting and boosting ensemble techniques is proposed. Ensemble methods are used to improve the predictive classification performance of the constituent classifiers by using multiple learning algorithms [
28]. Two ensemble techniques are used, voting, where several base models are trained and the decision is made based on the votes of each estimator, and boosting, which is mainly used to reduce bias and variance in order to make the estimators stronger. The proposed method adapts a heterogeneous voting approach using two base learners, random forest and support vector machine. The classification probabilities of the these base learners are combined to develop a voting ensemble technique based on the majority vote of class probability to obtain the final accuracy for the ensemble approach. In training each base learner, a boost approach is employed to improve the learners’ accuracy and reduce the false prediction rate. To the best of our knowledge, very few prior studies in gene expression analysis have tried to employ an over-sampling technique to handle the class imbalance problem. Therefore, in our approach’s boosting step, each learner is trained in a balanced subset from the samples, selected from the original majority class and the over-sampling produced for the minority class. The proposed approach performance is evaluated and compared with the base learner and one of the most recently published ensemble algorithms on five KEEL imbalanced datasets, as well as the MS expression data [
29]. For MS diagnosis, the experiments are carried out to evaluate the methods’ performance with all feature sets, differentially expressed genes, and reduced feature subsets from three Feature Selection (FS) algorithms. The FS algorithms employed are the Chi-squared algorithm [
30], as one of the most used algorithms for feature selection, Recursive Feature Elimination with Support Vector Clustering (RFE-SVC), as a gold standard wrapper algorithm that outweighs other FS algorithms [
31], the Extreme Gradient Boosting (XGBoost) for feature selection (BoostFS) [
32], and DEGs generated from the linear model of the Limma package [
33]. Limma is an R package developed for microarray data differential expression analysis. Moreover, Gene Ontology and KEGG Pathway enrichment analysis are performed for the identified DEGs, using the EnrichR 2.0 package [
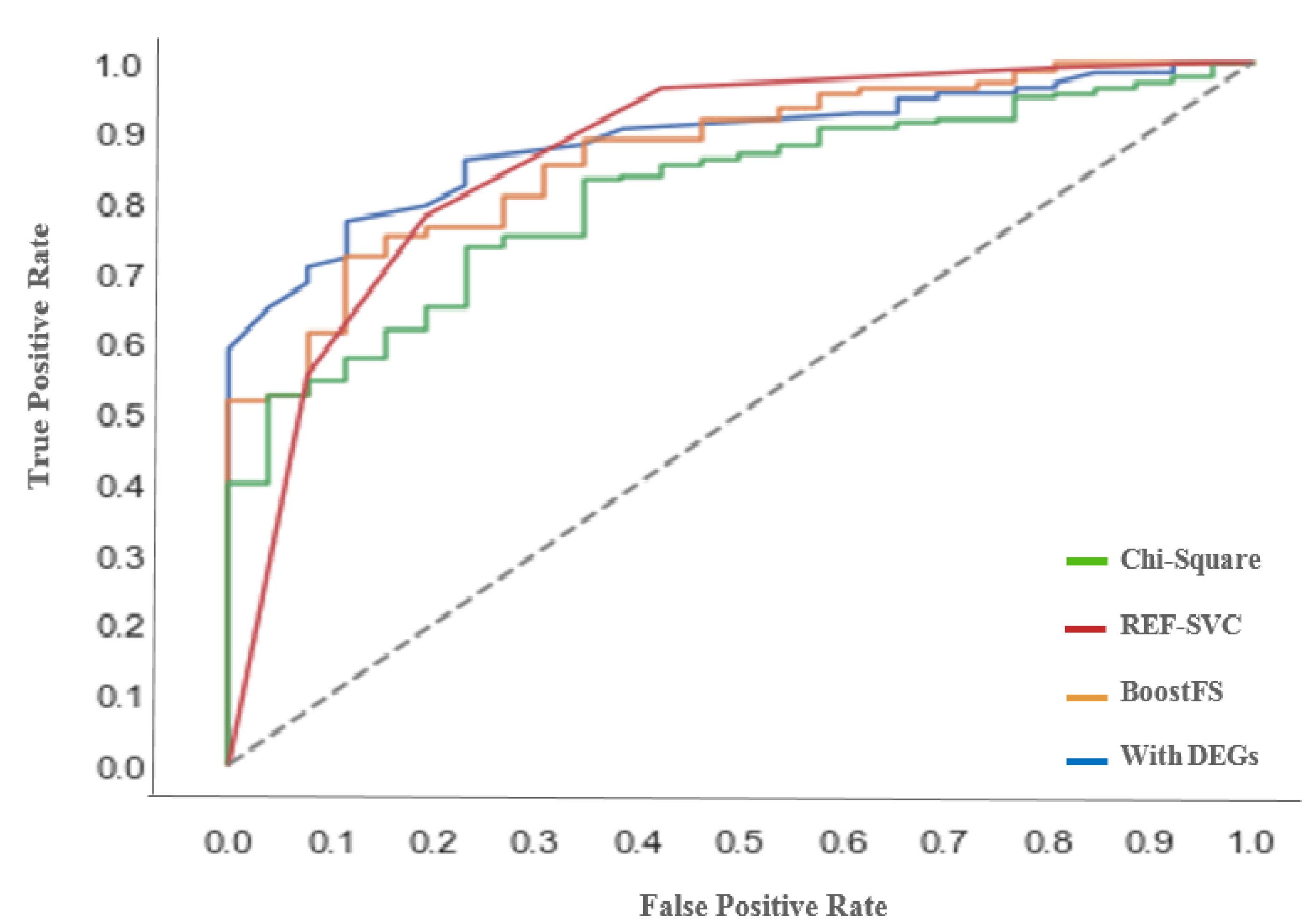
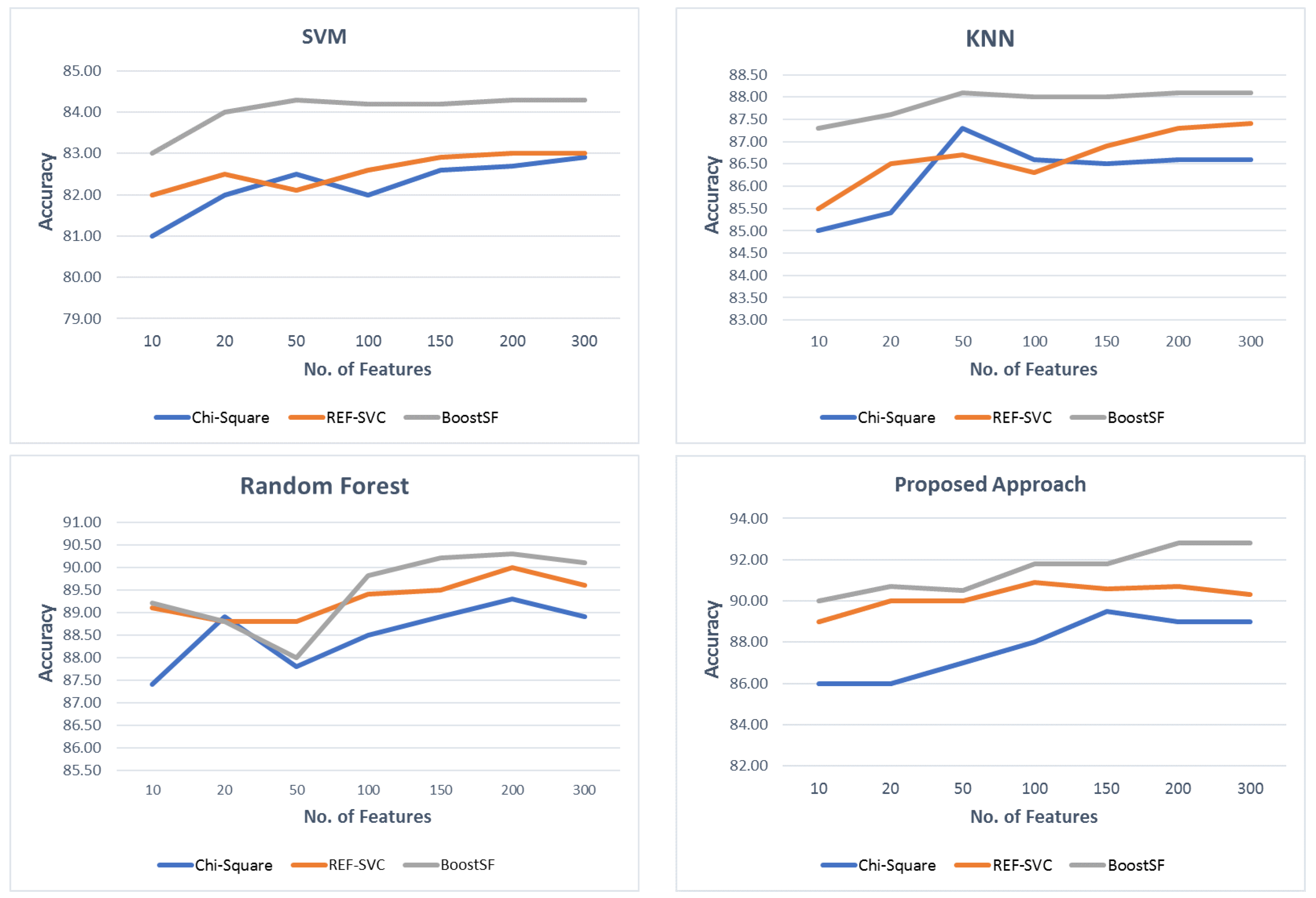
34] to reveal their functional relation to MS pathways. Accuracy, the Matthew Correlation Coefficient (MCC), Root-Mean-Squared Error (RMSE), F-score, Receiver Operating Characteristic (ROC) curve, and Area Under the Curve (AUC) are used as the evaluation metrics. Experimental results show that the proposed method achieves the highest accuracy of 92.81% and 91.8% with BoostFS and DEGs, respectively.
The contributions of this paper are summarized in the following points:
Proposing a hierarchical ensemble approach to improve the classification accuracy for multiple sclerosis patients.
Handling the class imbalance problem using over-sampling to improve the classifier performance when predicting the minority class.
Applying three different feature selection algorithms, in addition to selecting genes using bioinformatics analysis on differentially expressed genes, to select relevant genes based on their importance to overcome the low number of samples compared to the number of features (microarray genes set).
Identifying the differentially expressed genes between normal and multiple sclerosis patients’ expression profiles to use as one of the selected feature sets for evaluating our proposed approach.
Evaluating the proposed ensemble approach to analyze the predictive accuracy on each selected feature set from the FS algorithms and DEGs. The evaluation metrics calculated are the accuracy, MCC, RMSE, F-score, AUC, and ROC curve.
The remainder of this paper is organized as follows.
Section 2 presents the proposed framework. Following the methods, all experiments, the dataset, and results are given in the Results
Section 3. Finally, a discussion, conclusions, and future directions are given in
Section 4.
4. Conclusions
This paper presented an ensemble approach using voting and boosting techniques for the prediction of multiple sclerosis patients using gene expression profiles. Two base learners, namely random forest and support vector machine, were employed with the voting and boosting techniques. Moreover, over-sampling was used to handle the class imbalance problem in the gene expression data. The proposed method for the class imbalance problem was evaluated on five KEEL imbalanced datasets, and the results obtained for the proposed over-sampling approach showed a higher classification accuracy than existing methods.
The proposed classification approach for MS was tested with different feature sets, namely all features, differentially expressed genes set, and reduced feature subsets using three feature selection algorithms: Chi-squared, recursive feature elimination with support vector regression, and Extreme Gradient Boosting (XGBoost) for feature selection (BoostFS), as well as DEGs. The proposed method was compared to existing classifiers, namely random forest, KNN, and SVM. Experimental results showed that the proposed method achieved the highest accuracy of 92.81% and 91.8% with BoostFS and DEGs, respectively, in the diagnosis of MS. Hence, the proposed method outperforms the classification accuracy, MCC, AUC, and f1-score of all existing techniques.
To the best of our knowledge, not much work in the literature has been directed towards the diagnosis of MS, especially using gene expression. Yet, the proposed approach outperforms the prediction accuracy reported in [
14,
15,
16,
17], where they reported an accuracy ranging from 68% to 95%, and not all methods concentrated on diagnosis, but some on the correlation between different variables. The proposed approach reported the highest accuracy of 92.81%, using a combination of DEGs and genes selected from recent wrapper selection methods, such as XGBoost, for a more accurate MS diagnosis. Gene expression profiles pose a challenge for ML methods, as their high-dimensional data could lead to ML algorithm over-fitting. Furthermore, most gene expression profiles’ data suffer from a class imbalance, which adds more obstacles to the learning algorithm. In this paper, we employed an over-sampling technique to solve the imbalance problem. This study could be further expanded by proposing a novel over-sampling method and experimenting on more base classifiers for our ensemble approach. Furthermore, another direction could involve deeply mining the expression data of multiple sclerosis.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}