
Detection of Novel Biallelic Causative Variants in COL7A1 Gene by Whole-Exome Sequencing, Resulting in Congenital Recessive Dystrophic Epidermolysis Bullosa in Three Unrelated Families


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
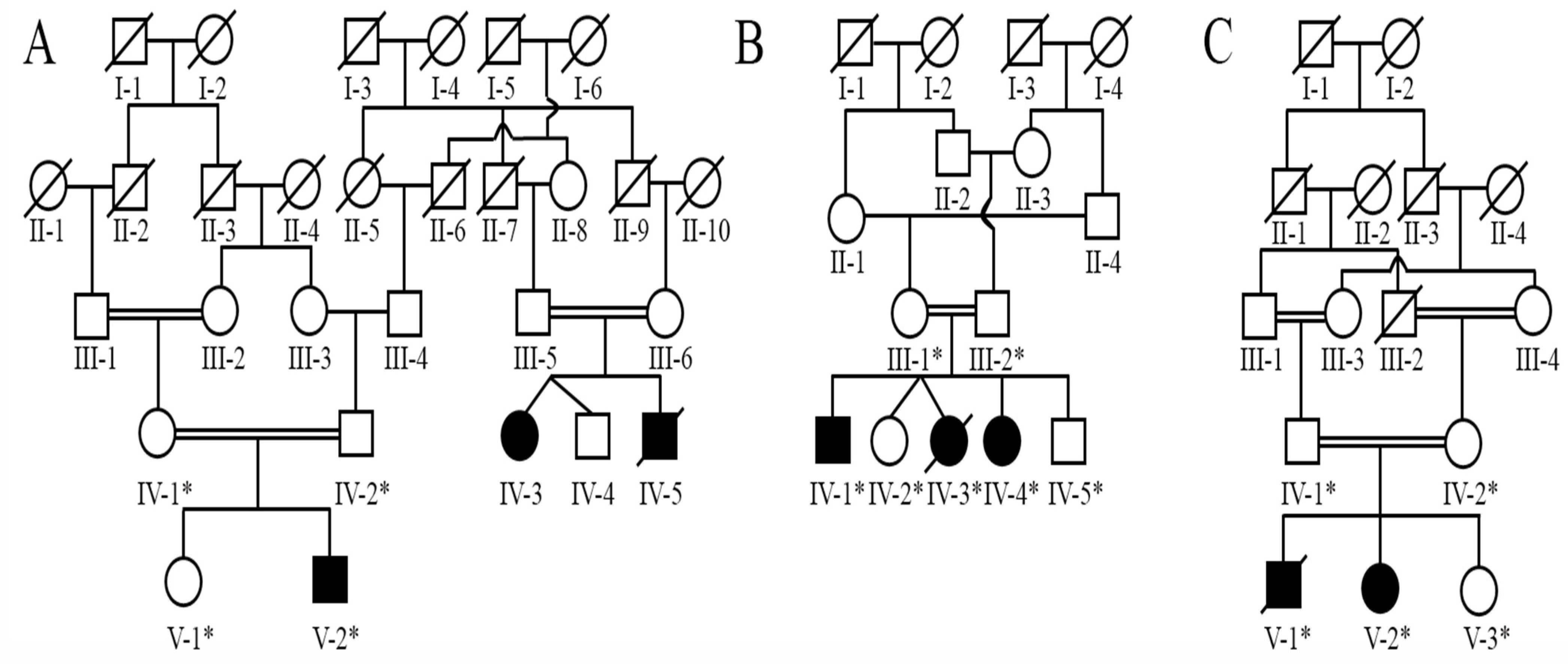
2.2. Family Recruitment
2.3. Blood Sample Collection
2.4. Genomic DNA Extraction
2.5. Whole-Exome Sequencing and Segregation of Rare Variants through Sanger Sequencing
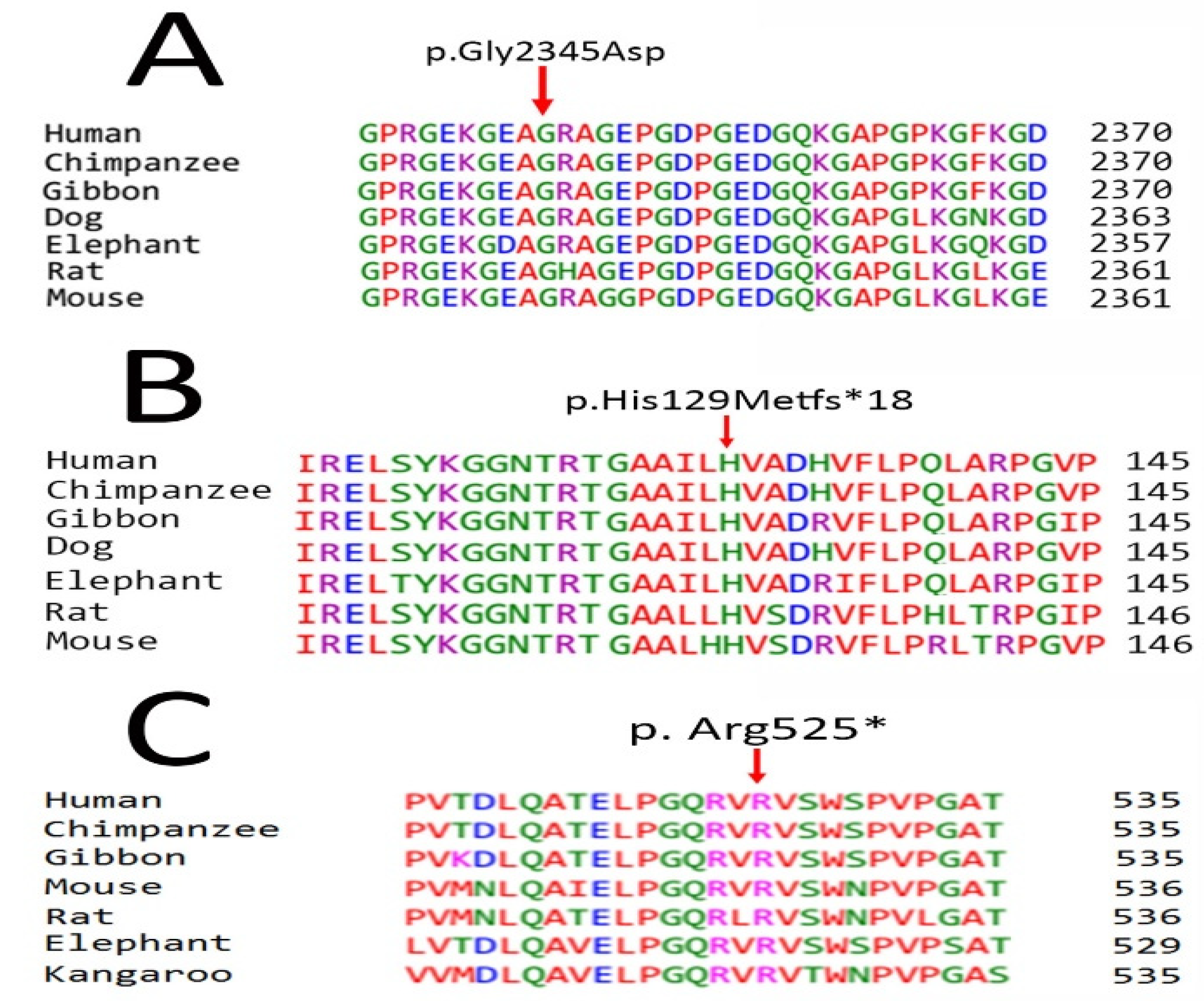
Pathogenicity Prediction and Protein Sequence Alignment
3. Results
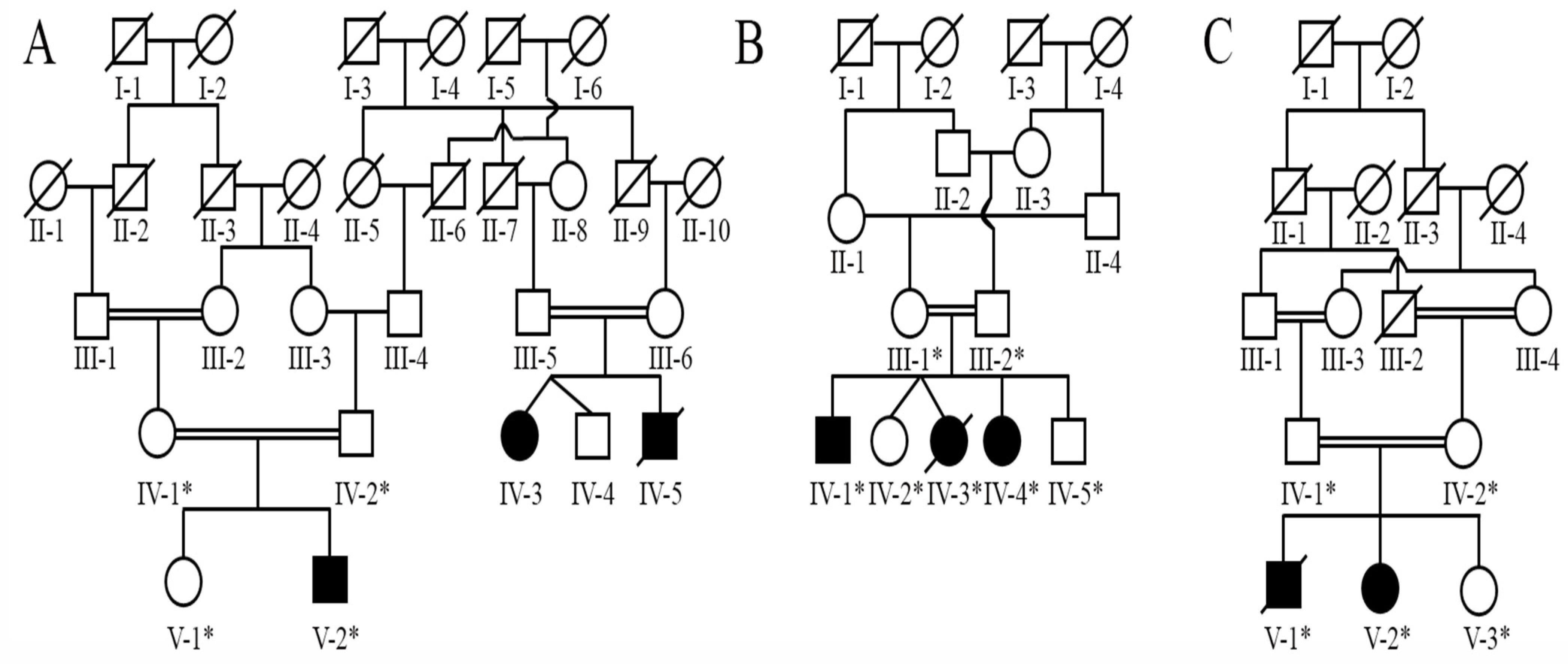
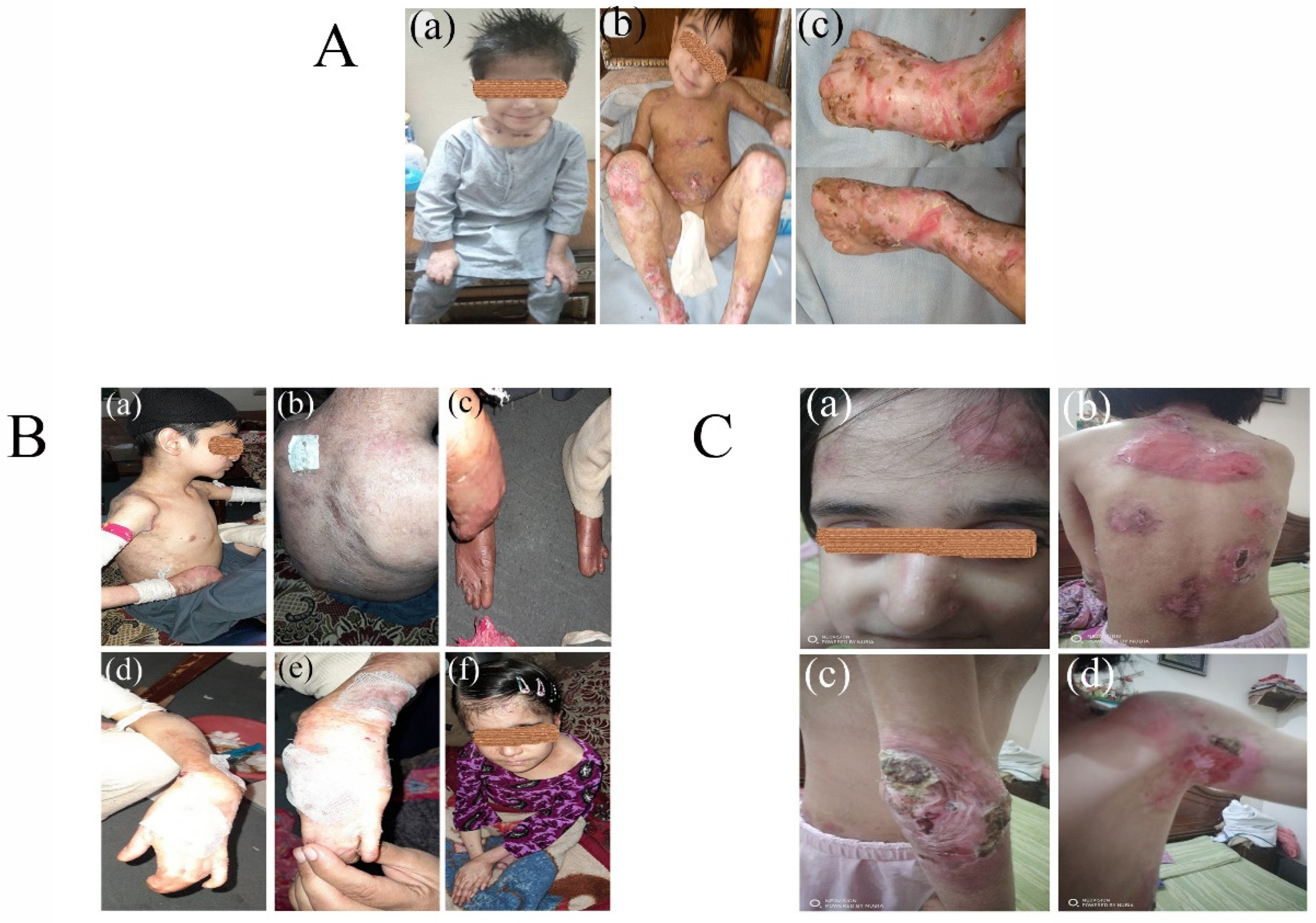
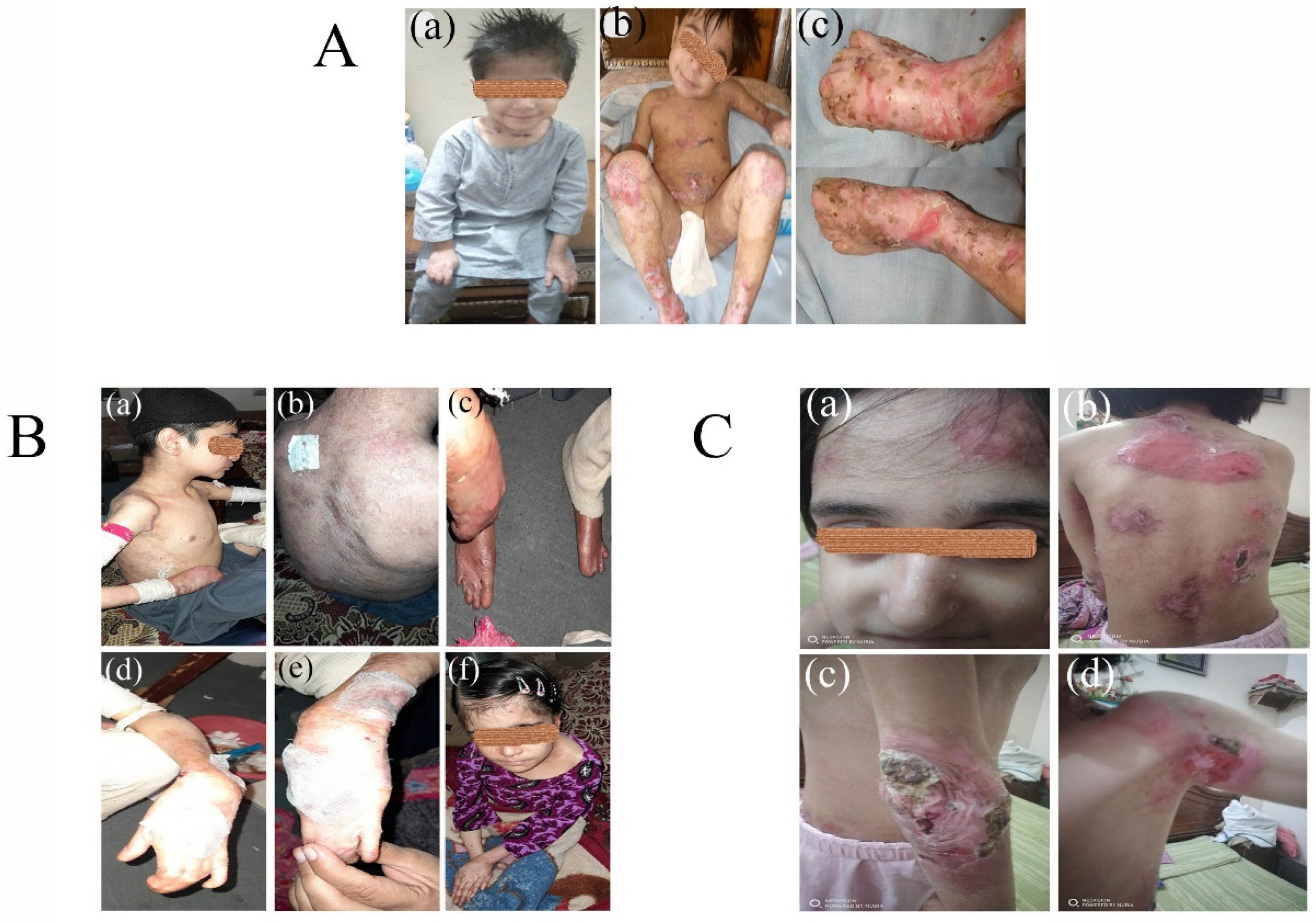
3.1. Clinical Phenotypes
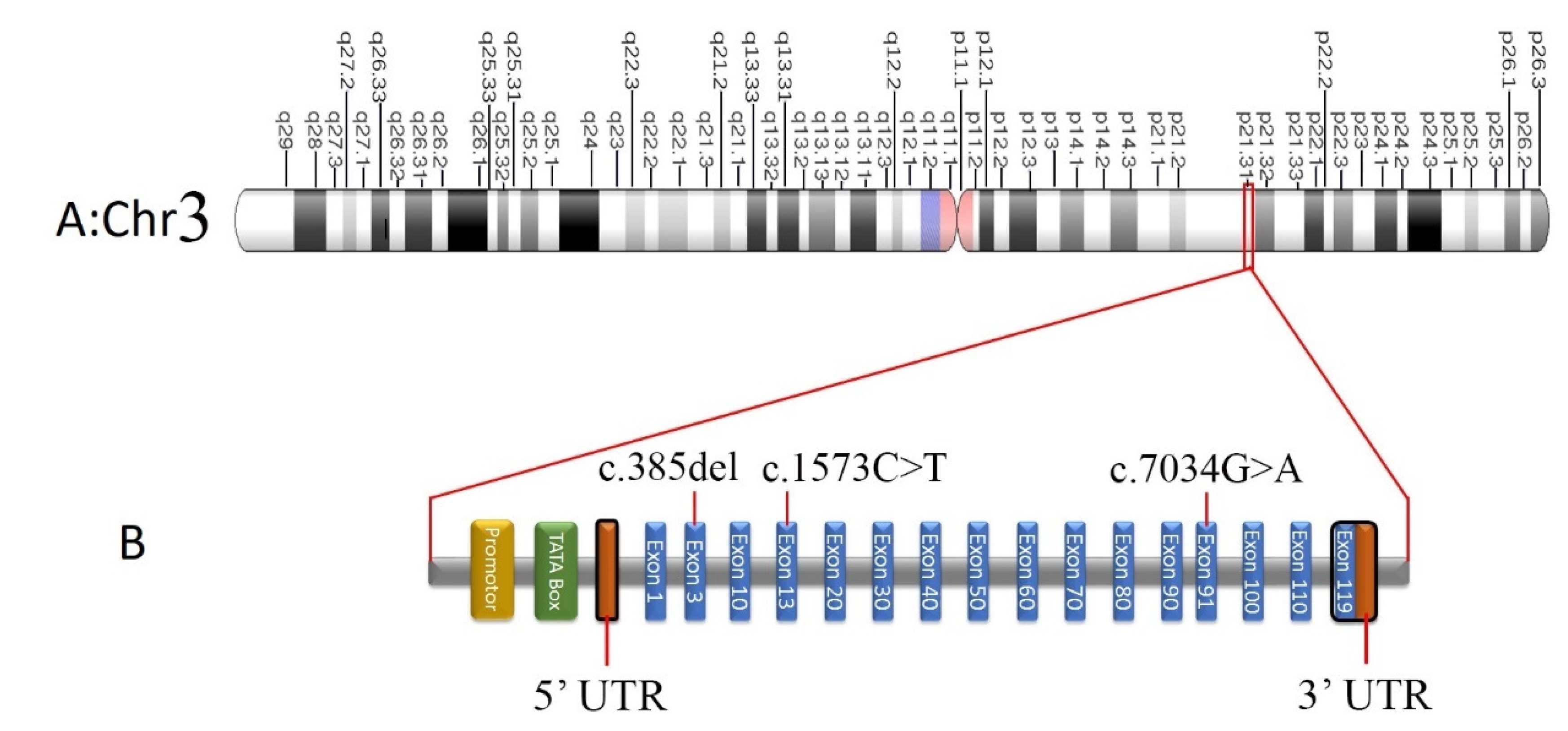
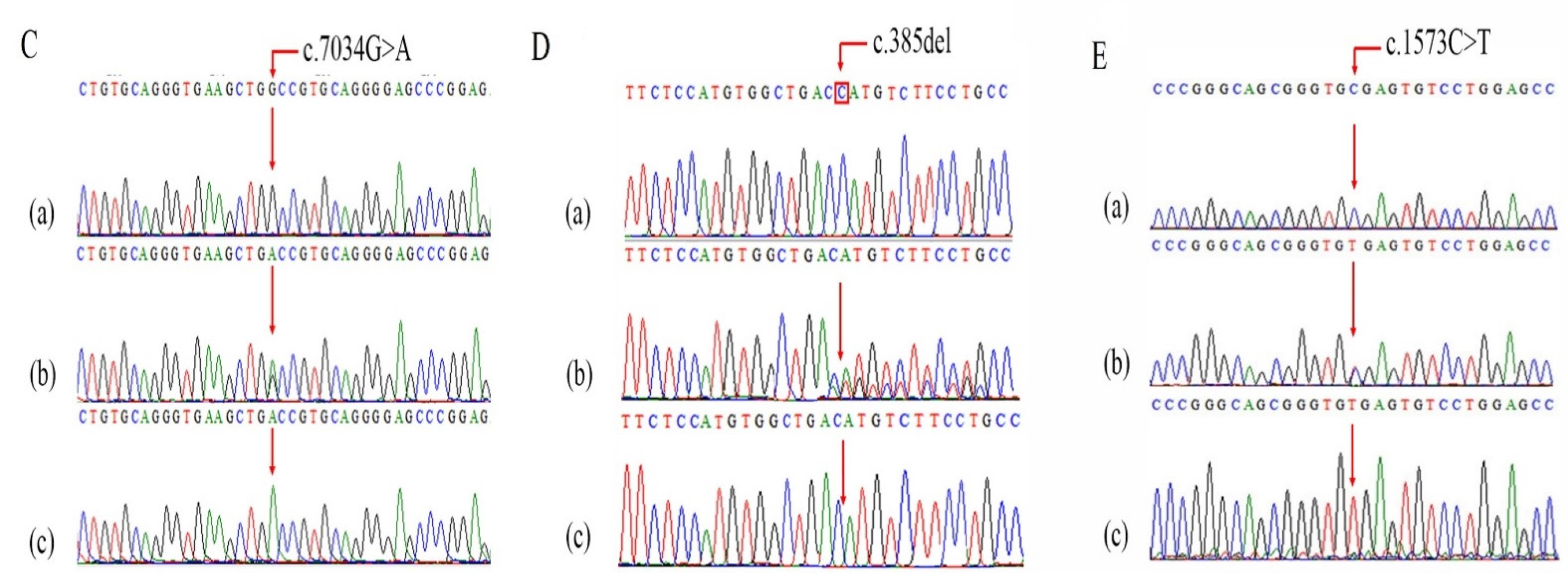
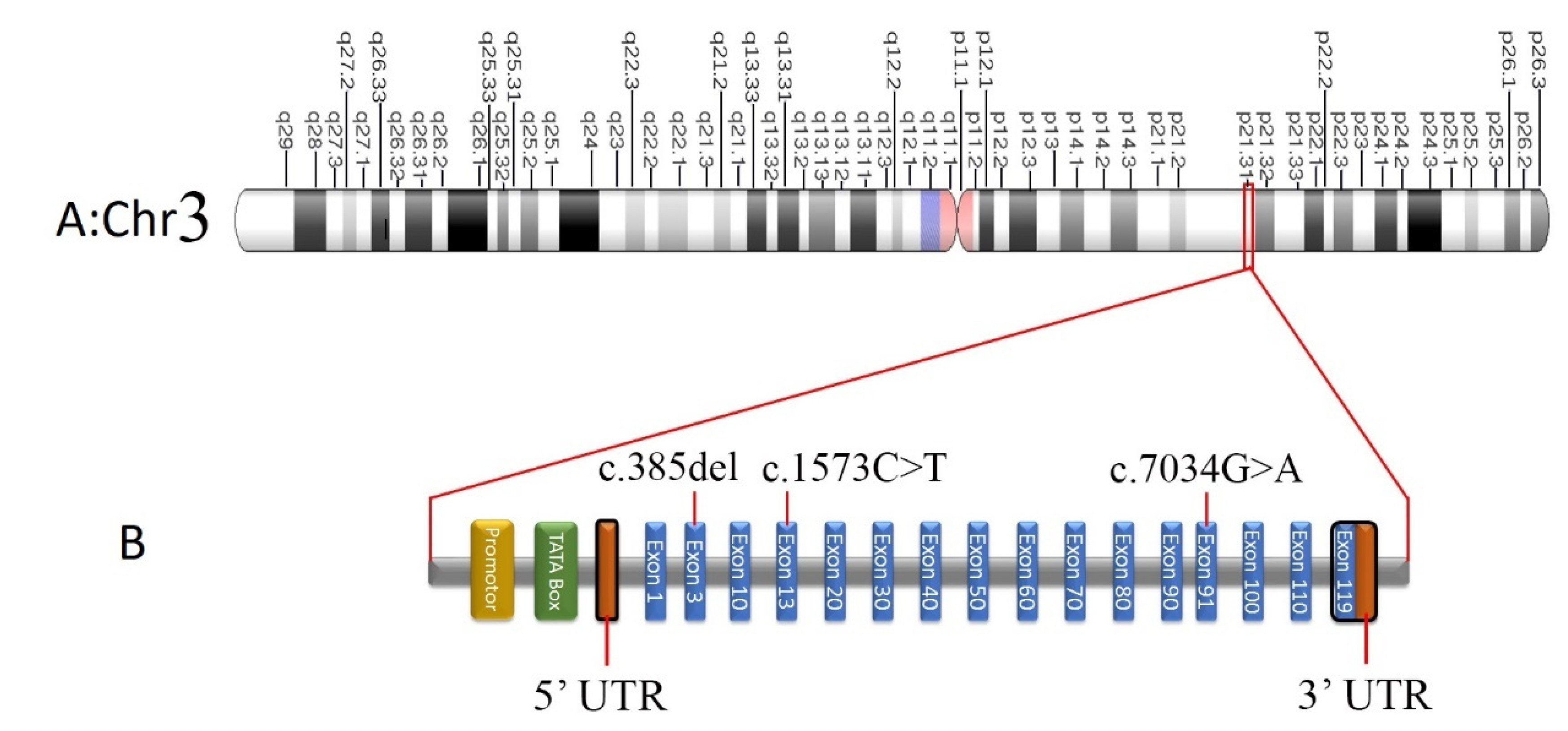
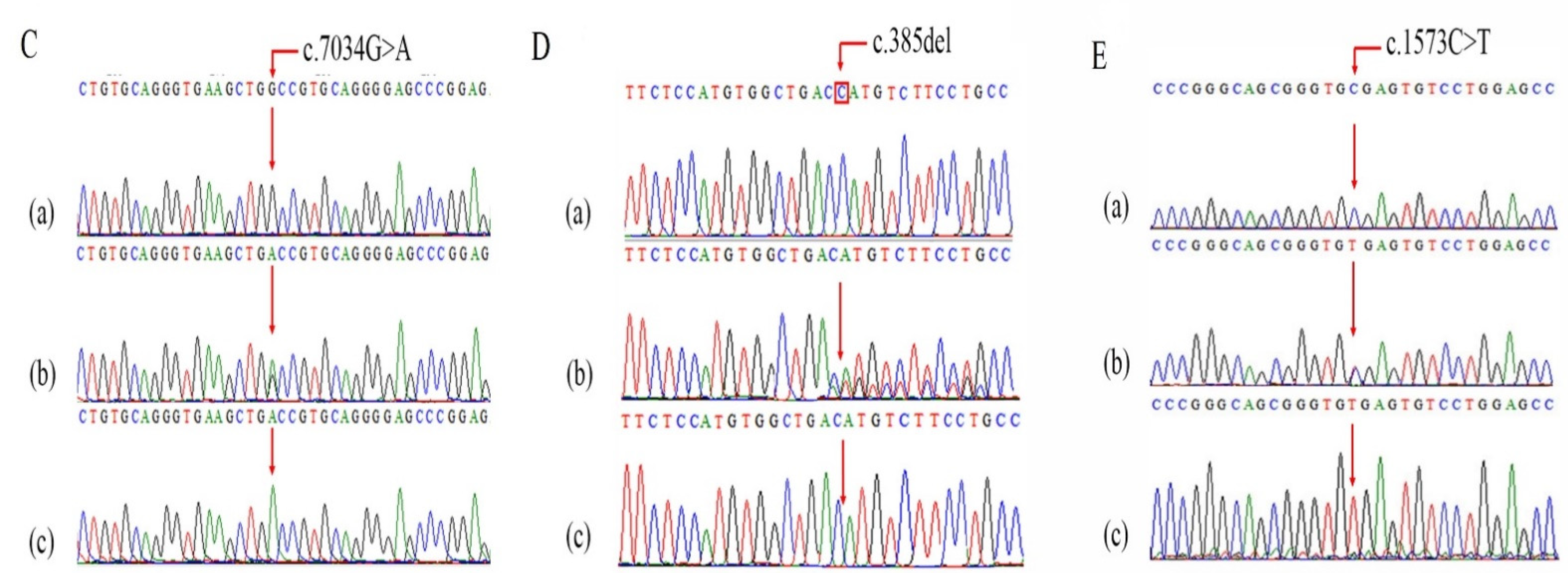
Screening of Pathogenic Sequence Variants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Chr. | Chr. Position | Variant Type | Cyto Band | GenBank | Ref. Seq. | Alt. Seq. | RS ID/dbSNP | UniProt ID | MAF gnomAD | MAF gnomAD South Asian |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COL7A1 | c.7034G>A; p.Gly2345Asp | 3 | 48609468 | SNV | 3p21.31 | NM_000094.4 | C | T | NR | Q02388 | NR | NR |
| COL7A1 | c.385delG; p.His129MetfsTer18 | 3 | 48631011 | Deletion | 3p21.31 | NM_000094.4 | G | - | NR | Q02388 | NR | NR |
| COL7A1 | c.1573C>T p.R525X | 3 | 48628960 | SNV | 3p21.31 | NM_000094.4 | G | A | rs368007918 | Q02388 | 0.00000796 | 0.0000327 |
| Gene | Variant | MT | PolyPhen2 | MUPRO | Fathmm | VarSome | PROVEAN | MutPred | SIFT |
|---|---|---|---|---|---|---|---|---|---|
| COL7A1 | c.7034G>A; p.Gly2345Asp | Disease causing | Possibly Damaging | Decrease stability | Damaging | Likely pathogenic | Deleterious | Pathogenic | Damaging |
| COL7A1 | c.385delG; p.His129MetfsTer18 | Disease causing | NA | NA | NA | Pathogenic | NA | NA | NA |
| COL7A1 | c.1573C>T p.R525X | Disease causing | NA | NA | Neutral | Pathogenic | NA | NA | NA |
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soro, L.; Bartus, C.; Purcell, S. Recessive dystrophic epidermolysis bullosa: A review of disease pathogenesis and update on future therapies. J. Clin. Aesthetic Dermatol. 2015, 8, 41–46. [Google Scholar]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Li, M.; Intong-Wheeler, L.R.A.; Tran, K.; Marucci, D.; Murrell, D.F. Epidemiology and Outcome of Squamous Cell Carcinoma in Epidermolysis Bullosa in Australia and New Zealand. Acta Derm. Venereol. 2018, 98, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidghamat, E.; McGrath, J.A. Novel and emerging therapies in the treatment of recessive dystrophic epidermolysis bullosa. Intractable Rare Dis. Res. 2017, 6, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denyer, J.; Pillay, E. Best practice guidelines for skin and wound care in epidermolysis bullosa: An international consensus. Wounds Int. 2017, 1, 7–29. [Google Scholar]
- Liy-Wong, C.A.A.; Aujla, N.; Briggs, L.; Campbell, F.; El Hamouly, A.; Section of Dermatology at the Hospital for Sick. Epidermolysis Bullosa, a Handbook for EB Patients and Families; DEBRA: Toronto, ON, Canada, 2021. [Google Scholar]
- Pfendner, E.G.; Lucky, A.W. Dystrophic Epidermolysis Bullosa. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Koch, L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 2020, 21, 448. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD(®)): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Pérez-Palma, E.; Gramm, M.; Nürnberg, P.; May, P.; Lal, D. Simple ClinVar: An interactive web server to explore and retrieve gene and disease variants aggregated in ClinVar database. Nucleic Acids Res. 2019, 47, W99–W105. [Google Scholar] [CrossRef] [Green Version]
- Qasim, I.; Ahmad, B.; Khan, M.A.; Khan, N.; Muhammad, N.; Basit, S.; Khan, S. Pakistan Genetic Mutation Database (PGMD); A centralized Pakistani mutome data source. Eur. J. Med. Genet. 2018, 61, 204–208. [Google Scholar] [CrossRef]
- Varki, R.; Sadowski, S.; Uitto, J.; Pfendner, E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J. Med. Genet. 2007, 44, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Christiano, A.M.; Anhalt, G.; Gibbons, S.; Bauer, E.A.; Uitto, J. Premature termination codons in the type VII collagen gene (COL7A1) underlie severe, mutilating recessive dystrophic epidermolysis bullosa. Genomics 1994, 21, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L. Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix Bio. 2018, 68–69, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Castiglia, D.; Zambruno, G. Molecular testing in epidermolysis bullosa. Dermatol. Clin. 2010, 28, 223–229, vii–viii. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bösl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckner-Tuderman, L. Dystrophic epidermolysis bullosa: Pathogenesis and clinical features. Dermatol. Clin. 2010, 28, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [Green Version]
- Bonamonte, D.; Filoni, A.; De Marco, A.; Lospalluti, L.; Nacchiero, E.; Ronghi, V.; Colagrande, A.; Giudice, G.; Cazzato, G. Squamous Cell Carcinoma in Patients with Inherited Epidermolysis Bullosa: Review of Current Literature. Cells 2022, 11, 1365. [Google Scholar] [CrossRef]
- Intong, L.R.; Murrell, D.F. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin. Dermatol. 2012, 30, 70–77. [Google Scholar] [CrossRef]
- Filoni, A.; Cicco, G.; Cazzato, G.; Bosco, A.; Lospalluti, L.; Tucci, M.; Cimmino, A.; Foti, C.; Marzullo, A.; Bonamonte, D. Immune Disregulation in Cutaneous Squamous Cell Carcinoma of Patients with Recessive Dystrophic Epidermolysis Bullosa: A Single Pilot Study. Life 2022, 12, 213. [Google Scholar] [CrossRef]
- March, O.P.; Reichelt, J.; Koller, U. Gene editing for skin diseases: Designer nucleases as tools for gene therapy of skin fragility disorders. Exp. Physiol. 2018, 103, 449–455. [Google Scholar] [CrossRef] [Green Version]
- March, O.P.; Kocher, T.; Koller, U. Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells 2020, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aushev, M.; Koller, U.; Mussolino, C.; Cathomen, T.; Reichelt, J. Traceless Targeting and Isolation of Gene-Edited Immortalized Keratinocytes from Epidermolysis Bullosa Simplex Patients. Mol. Ther. Methods Clin. Dev. 2017, 6, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, O.P.; Lettner, T.; Klausegger, A.; Ablinger, M.; Kocher, T.; Hainzl, S.; Peking, P.; Lackner, N.; Rajan, N.; Hofbauer, J.P.; et al. Gene Editing–Mediated Disruption of Epidermolytic Ichthyosis–Associated KRT10 Alleles Restores Filament Stability in Keratinocytes. J. Investig. Dermatol. 2019, 139, 1699–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, C.; Mencía, A.; Almarza, D.; Duarte, B.; Büning, H.; Sallach, J.; Hausser, I.; Del Río, M.; Larcher, F.; Murillas, R. Gene Editing for the Efficient Correction of a Recurrent COL7A1 Mutation in Recessive Dystrophic Epidermolysis Bullosa Keratinocytes. Mol. Ther. Nucleic Acids 2016, 5, e307. [Google Scholar] [CrossRef] [Green Version]
- Mencía, Á.; Chamorro, C.; Bonafont, J.; Duarte, B.; Holguin, A.; Illera, N.; Llames, S.G.; Escámez, M.J.; Hausser, I.; Del Río, M.; et al. Deletion of a Pathogenic Mutation-Containing Exon of COL7A1 Allows Clonal Gene Editing Correction of RDEB Patient Epidermal Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Takashima, S.; Shinkuma, S.; Fujita, Y.; Nomura, T.; Ujiie, H.; Natsuga, K.; Iwata, H.; Nakamura, H.; Vorobyev, A.; Abe, R.; et al. Efficient Gene Reframing Therapy for Recessive Dystrophic Epidermolysis Bullosa with CRISPR/Cas9. J. Investig. Dermatol. 2019, 139, 1711–1721. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Chacón-Solano, E.; Modamio-Høybjør, S.; Marinas, L.; León, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef]
- Chakrabarti, A.M.; Henser-Brownhill, T.; Monserrat, J.; Poetsch, A.R.; Luscombe, N.M.; Scaffidi, P. Target-Specific Precision of CRISPR-Mediated Genome Editing. Mol. Cell 2019, 73, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Kocher, T.; March, O.P.; Bischof, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; Hoog, A.; Strunk, D.; Bauer, J.W.; Koller, U. Predictable CRISPR/Cas9-Mediated COL7A1 Reframing for Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2020, 140, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E.; Ablinger, M.; Lettner, T.; Murauer, E.M.; Guttmann-Gruber, C.; Piñón Hofbauer, J.; Hainzl, S.; Kaiser, M.; Klausegger, A.; Bauer, J.W.; et al. 5’RNA Trans-Splicing Repair of COL7A1 Mutant Transcripts in Epidermolysis Bullosa. Int. J. Mol. Sci. 2022, 23, 1732. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part I. Epithelial associated tissues. J. Am. Acad. Dermatol. 2009, 61, 366–385. [Google Scholar] [CrossRef]
- Horn, H.M.; Tidman, M.J. The clinical spectrum of dystrophic epidermolysis bullosa. Br. J. Dermatol. 2002, 146, 267–274. [Google Scholar] [CrossRef]
- Tong, L.; Hodgkins, P.R.; Denyer, J.; Brosnahan, D.; Harper, J.; Russell-Eggitt, I.; Taylor, D.S.; Atherton, D. The eye in epidermolysis bullosa. Br. J. Ophthalmol. 1999, 83, 323–326. [Google Scholar] [CrossRef]
- Mellado, F.; Fuentes, I.; Palisson, F.; Vergara, J.I.; Kantor, A. Ophthalmologic Approach in Epidermolysis Bullosa: A Cross-Sectional Study With Phenotype-Genotype Correlations. Cornea 2018, 37, 442–447. [Google Scholar] [CrossRef]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M.G. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Kiritsi, D.; Nyström, A. Recent advances in understanding and managing epidermolysis bullosa. F1000Research 2018, 7, 1097. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fozia, F.; Nazli, R.; Alrashed, M.M.; Ghneim, H.K.; Haq, Z.U.; Jabeen, M.; Alam Khan, S.; Ahmad, I.; Bourhia, M.; Aboul-Soud, M.A.M. Detection of Novel Biallelic Causative Variants in COL7A1 Gene by Whole-Exome Sequencing, Resulting in Congenital Recessive Dystrophic Epidermolysis Bullosa in Three Unrelated Families. Diagnostics 2022, 12, 1525. https://doi.org/10.3390/diagnostics12071525
Fozia F, Nazli R, Alrashed MM, Ghneim HK, Haq ZU, Jabeen M, Alam Khan S, Ahmad I, Bourhia M, Aboul-Soud MAM. Detection of Novel Biallelic Causative Variants in COL7A1 Gene by Whole-Exome Sequencing, Resulting in Congenital Recessive Dystrophic Epidermolysis Bullosa in Three Unrelated Families. Diagnostics. 2022; 12(7):1525. https://doi.org/10.3390/diagnostics12071525
Chicago/Turabian StyleFozia, Fozia, Rubina Nazli, May Mohammed Alrashed, Hazem K. Ghneim, Zia Ul Haq, Musarrat Jabeen, Sher Alam Khan, Ijaz Ahmad, Mohammed Bourhia, and Mourad A. M. Aboul-Soud. 2022. "Detection of Novel Biallelic Causative Variants in COL7A1 Gene by Whole-Exome Sequencing, Resulting in Congenital Recessive Dystrophic Epidermolysis Bullosa in Three Unrelated Families" Diagnostics 12, no. 7: 1525. https://doi.org/10.3390/diagnostics12071525
APA StyleFozia, F., Nazli, R., Alrashed, M. M., Ghneim, H. K., Haq, Z. U., Jabeen, M., Alam Khan, S., Ahmad, I., Bourhia, M., & Aboul-Soud, M. A. M. (2022). Detection of Novel Biallelic Causative Variants in COL7A1 Gene by Whole-Exome Sequencing, Resulting in Congenital Recessive Dystrophic Epidermolysis Bullosa in Three Unrelated Families. Diagnostics, 12(7), 1525. https://doi.org/10.3390/diagnostics12071525