
Molecular Landscape of Pediatric Thyroid Cancer: A Review



Abstract
:1. Introduction
2. Epidemiology
3. Risk Factors
3.1. Exposure to Radiation/Radiotherapy
3.2. Autoimmune Thyroiditis
3.3. Iodine Content
3.4. Familial/Genetic Syndromes
4. Molecular Profile of Differentiated Thyroid Carcinomas
4.1. Papillary Thyroid Carcinoma
4.2. Follicular Thyroid Carcinoma
4.3. Poorly Differentiated Thyroid Carcinoma
4.4. Medullary Thyroid Carcinoma
5. Role of miRNA in Pediatric Thyroid Carcinoma
6. Other Potential Biomolecules
7. Targeted Therapy in Pediatric Patients
8. Molecular Evaluation of Indeterminate Thyroid Nodules in Pediatric Patients
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, A.J. Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol. Metab. Clin. N. Am. 2017, 46, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.D.; Pantanowitz, L.; Hornick, J.L. A worldwide journey of thyroid cancer incidence centred on tumour histology. Lancet Diabetes Endocrinol. 2021, 9, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Cancer Statistics Review, 1975–2018—SEER Statistics. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 14 January 2022).
- Chan, C.M.; Young, J.; Prager, J.; Travers, S. Pediatric Thyroid Cancer. Adv. Pediatr. 2017, 64, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Adolescent Health. Available online: https://www.who.int/southeastasia/health-topics/adolescent-health (accessed on 14 December 2021).
- Francis, G.L.; Waguespack, S.G.; Bauer, A.J.; Angelos, P.; Benvenga, S.; Cerutti, J.M.; Dinauer, C.A.; Hamilton, J.; Hay, I.D.; Luster, M.; et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015, 25, 716–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, A.S.; Qasem, E.; Murugan, A.K.; Al-Hindi, H.N.; Alkhafaji, D.; Almohanna, M.; Xing, M.; Alhomaidah, D.; Alswailem, M. Uncommon TERT promoter mutations in pediatric thyroid cancer. Thyroid 2016, 26, 235–241. [Google Scholar] [CrossRef]
- Hardin, A.P.; Hackell, J.M.; Simon, G.R.; Boudreau, A.D.A.; Baker, C.N.; Barden, G.A.; Meade, K.E.; Moore, S.B.; Richerson, J.; Brown, O.W.; et al. Age Limit of Pediatrics. Pediatrics 2017, 140, e20172151. [Google Scholar] [CrossRef] [Green Version]
- Buryk, M.A.; Simons, J.P.; Picarsic, J.; Monaco, S.E.; Ozolek, J.A.; Joyce, J.; Gurtunca, N.; Nikiforov, Y.E.; Witchel, S.F. Can malignant thyroid nodules be distinguished from benign thyroid nodules in children and adolescents by clinical characteristics? A review of 89 pediatric patients with thyroid nodules. Thyroid 2015, 25, 392–400. [Google Scholar] [CrossRef]
- Decaussin-Petrucci, M.; Deladoëy, J.; Hafdi-Nejjari, Z.; Sassolas, G.; Borson-Chazot, F.; Abu-Khudir, R.; Fusco, A.; Descotes, F.; Cournoyer, S.; Sartelet, H.; et al. Expression of CD133 in differentiated thyroid cancer of young patients. J. Clin. Pathol. 2015, 68, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Espadinha, C.; Santos, J.R.; Sobrinho, L.G.; Bugalho, M.J. Expression of iodine metabolism genes in human thyroid tissues: Evidence for age and BRAF V600E mutation dependency. Clin. Endocrinol. 2009, 70, 629–635. [Google Scholar] [CrossRef]
- Monaco, S.E.; Pantanowitz, L.; Khalbuss, W.E.; Benkovich, V.A.; Ozolek, J.; Nikiforova, M.N.; Simons, J.P.; Nikiforov, Y.E. Cytomorphological and molecular genetic findings in pediatric thyroid fine-needle aspiration. Cancer Cytopathol. 2012, 120, 342–350. [Google Scholar] [CrossRef]
- Sugino, K.; Nagahama, M.; Kitagawa, W.; Ohkuwa, K.; Matsuzu, K.; Suzuki, A.; Tomoda, C.; Hames, K.Y.; Akaishi, J.; Masaki, C.; et al. Cutoff Age Between Pediatric and Adult Thyroid Differentiated Cancer: Is 18 Years Old Appropriate? Thyroid 2021, 32, 145–152. [Google Scholar] [CrossRef]
- Paulson, V.A.; Rudzinski, E.R.; Hawkins, D.S. Thyroid cancer in the pediatric population. Genes 2019, 10, 723. [Google Scholar] [CrossRef] [Green Version]
- Cherella, C.E.; Angell, T.E.; Richman, D.M.; Frates, M.C.; Benson, C.B.; Moore, F.D.; Barletta, J.A.; Hollowell, M.; Smith, J.R.; Alexander, E.K.; et al. Differences in Thyroid Nodule Cytology and Malignancy Risk Between Children and Adults. Thyroid 2019, 29, 1097–1104. [Google Scholar] [CrossRef]
- Qian, Z.J.; Jin, M.C.; Meister, K.D.; Megwalu, U.C. Pediatric Thyroid Cancer Incidence and Mortality Trends in the United States, 1973–2013. JAMA Otolaryngol.—Head Neck Surg. 2019, 145, 617–623. [Google Scholar] [CrossRef]
- Jarząb, B.; Handkiewicz-Junak, D.; Włoch, J. Juvenile differentiated thyroid carcinoma and the role of radioiodine in its treatment: A qualitative review. Endocr. Relat. Cancer 2005, 12, 773–803. [Google Scholar] [CrossRef]
- Hogan, A.R.; Zhuge, Y.; Perez, E.A.; Koniaris, L.G.; Lew, J.I.; Sola, J.E. Pediatric thyroid carcinoma: Incidence and outcomes in 1753 patients. J. Surg. Res. 2009, 156, 167–172. [Google Scholar] [CrossRef]
- Demidchik, Y.E.; Saenko, V.A.; Yamashita, S. Childhood thyroid cancer in Belarus, Russia, and Ukraine after Chernobyl and at present. Arq. Bras. Endocrinol. Metabol. 2007, 51, 748–762. [Google Scholar] [CrossRef] [Green Version]
- Cancer Today. Available online: https://gco.iarc.fr/today/online-analysis-pie?v=2020&mode=population&mode_population=continents&population=900&populations=900&key=total&sex=0&cancer=32&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=3&nb_items=7&group_cancer=0&include_nmsc=1&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 22 January 2022).
- Parisi, M.T.; Eslamy, H.; Mankoff, D. Management of differentiated thyroid cancer in children: Focus on the American Thyroid Association pediatric guidelines. Semin. Nucl. Med. 2016, 46, 147–164. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.; Fagin, J.A. Prevalence of minisatellite and microsatellite instability in radiation-induced post-Chernobyl pediatric thyroid carcinomas. Oncogene 1998, 17, 1983–1988. [Google Scholar] [CrossRef] [Green Version]
- Niedziela, M. Pathogenesis, diagnosis and management of thyroid nodules in children. Endocr. Relat. Cancer 2006, 13, 427–453. [Google Scholar] [CrossRef]
- Nikiforov, Y.; Gnepp, D.R.; Fagin, J.A. Thyroid lesions in children and adolescents after the Chernobyl disaster: Implications for the study of radiation tumorigenesis. J. Clin. Endocrinol. Metab. 1996, 81, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazakov, V.S.; Demidchik, E.P.; Astakhova, L.N.; Baverstock, K.; Egloff, B.; Pinchera, A.; Ruchti, C.; Williams, D. Thyroid cancer after Chernobyl; and reply. Nature 1992, 21, 359. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Rowland, J.M.; Bove, K.E.; Monforte-Munoz, H.; Fagin, J.A. Distinct Pattern of ret Oncogene Rearrangements in Morphological Variants of Radiation-induced and Sporadic Thyroid Papillary Carcinomas in Children. Cancer Res. 1997, 57, 1690–1694. [Google Scholar] [PubMed]
- Fenton, C.L.; Lukes, Y.; Nicholson, D.; Dinauer, C.A.; Francis, G.L.; Tuttle, R.M. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J. Clin. Endocrinol. Metab. 2000, 85, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Rabes, H.M.; Demidchik, E.P.; Sidorow, J.D.; Lengfelder, E.; Beimfohr, C.; Hoelzel, D.; Klugbauer, S. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: Biological, phenotypic, and clinical implications. Clin. Cancer Res. 2000, 6, 1093–1103. [Google Scholar]
- Himizu, K.; Tanaka, S.; Fujimori, M.; Yokoyama, S.; Ito, K.; Emi, M. Fusion of a novel gene, ELKS, to RET due to translocation t(10;12)(q11;p13) in a papillary thyroid carcinoma. Genes Chromosomes Cancer 1999, 25, 97–103. [Google Scholar]
- Iyama, K.; Matsuse, M.; Mitsutake, N.; Rogounovitch, T.; Saenko, V.; Suzuki, K.; Ashizawa, M.; Ookouchi, C.; Suzuki, S.; Mizunuma, H.; et al. Identification of Three Novel Fusion Oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in Thyroid Cancers of Young Patients in Fukushima. Thyroid 2017, 27, 811–818. [Google Scholar] [CrossRef]
- Dvorkin, S.; Robenshtok, E.; Hirsch, D.; Strenov, Y.; Shimon, I.; Benbassat, C.A. Differentiated thyroid cancer is associated with less aggressive disease and better outcome in patients with coexisting Hashimotos thyroiditis. J. Clin. Endocrinol. Metab. 2013, 98, 2409–2414. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.C.; Greenspan, F.S.; Dong, F.; Miller, T.R.; Yeo, P.P.B. Influence of lymphocytic thyroiditis on the prognostic outcome of patients with papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 1999, 84, 458–463. [Google Scholar] [CrossRef]
- Kashima, K.; Yokoyama, S.; Noguchi, S.; Murakami, N.; Yamashita, H.; Watanabe, S.; Uchino, S.; Toda, M.; Sasaki, A.; Daa, T.; et al. Chronic thyroiditis as a favorable prognostic factor in papillary thyroid carcinoma. Thyroid 1998, 8, 197–202. [Google Scholar] [CrossRef]
- Singh, B.; Shaha, A.R.; Trivedi, H.; Carew, J.F.; Poluri, A.; Shah, J.P. Coexistent Hashimoto’s thyroiditis with papillary thyroid carcinoma: Impact on presentation, management, and outcome. Surgery 1999, 126, 1070–1077. [Google Scholar] [CrossRef]
- Corrias, A.; Cassio, A.; Weber, G.; Mussa, A.; Wasniewska, M.; Rapa, A.; Gastaldi, R.; Einaudi, S.; Baronio, F.; Vigone, M.C.; et al. Thyroid Nodules and Cancer in Children and Adolescents Affected by Autoimmune Thyroiditis. Arch. Pediatr. Adolesc. Med. 2008, 162, 526–531. [Google Scholar] [CrossRef]
- Sur, M.L.; Gaga, R.; Lazăr, C.; Lazea, C.; Aldea, C.; Sur, D. Papillary thyroid carcinoma in children with Hashimoto’s thyroiditis—A review of the literature between 2000 and 2020. J. Pediatr. Endocrinol. Metab. 2020, 33, 1511–1517. [Google Scholar] [CrossRef]
- Ren, P.Y.; Liu, J.; Xue, S.; Chen, G. Pediatric differentiated thyroid carcinoma: The clinicopathological features and the coexistence of Hashimoto’s thyroiditis. Asian J. Surg. 2019, 42, 112–119. [Google Scholar] [CrossRef]
- Penta, L.; Cofini, M.; Lanciotti, L.; Leonardi, A.; Principi, N.; Esposito, S. Hashimoto’s Disease and Thyroid Cancer in Children: Are They Associated? Front. Endocrinol. 2018, 9, 565. [Google Scholar] [CrossRef] [Green Version]
- Subhi, O.; Schulten, H.J.; Bagatian, N.; Al-Dayini, R.; Karim, S.; Bakhashab, S.; Alotibi, R.; Al-Ahmadi, A.; Ata, M.; Elaimi, A.; et al. Genetic relationship between Hashimoto’s thyroiditis and papillary thyroid carcinoma with coexisting Hashimoto’s thyroiditis. PLoS ONE 2020, 15, e0234566. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Galetti, V. Iodine intake as a risk factor for thyroid cancer: A comprehensive review of animal and human studies. Thyroid Res. 2015, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Drozd, V.; Branovan, D.I.; Reiners, C. Increasing Incidence of Thyroid Carcinoma: Risk Factors and Seeking Approaches for Primary Prevention. Int. J. Thyroidol. 2020, 13, 95–110. [Google Scholar] [CrossRef]
- Kumar, A.; Bal, C.S. Differentiated thyroid cancer. Indian J. Pediatr. 2003, 70, 707–713. [Google Scholar] [CrossRef]
- Agarwal, S.; Bychkov, A.; Jung, C.-K. Emerging Biomarkers in Thyroid Practice and Research. Cancers 2021, 14, 204. [Google Scholar] [CrossRef]
- Nosé, V. Familial thyroid cancer: A review. Mod. Pathol. 2011, 24, S19–S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannelli, S.M.; McPhaul, L.; Nakamoto, J.; Gianoukakis, A.G. Familial adenomatous polyposis-associated, cribriform morular variant of papillary thyroid carcinoma harboring a K-RAS mutation: Case presentation and review of molecular mechanisms. Thyroid 2014, 24, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.L. Familial syndromes associated with thyroid cancer in the era of personalized medicine. Thyroid 2010, 20, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Jung, S.H.; Hirokawa, M.; Bychkov, A.; Miyauchi, A.; Lee, S.; Chung, Y.J.; Jung, C.K. High Prevalence of DICER1 Mutations and Low Frequency of Gene Fusions in Pediatric Follicular-Patterned Tumors of the Thyroid. Endocr. Pathol. 2021, 32, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, J.D.; Sabbaghian, N.; Fahiminiya, S.; Chami, R.; Mete, O.; Acker, M.; Wu, M.K.; Shlien, A.; De Kock, L.; Foulkes, W.D. DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 2009–2015. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.A.; Im, S.W.; Jung, K.C.; Chung, E.J.; Shin, C.H.; Kim, J.I.; Park, Y.J. Predominant DICER1 Pathogenic Variants in Pediatric Follicular Thyroid Carcinomas. Thyroid 2020, 30, 1120–1131. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Stenman, A.; Zedenius, J. Macrofollicular variant follicular thyroid tumors are DICER1 mutated and exhibit distinct histological features. Histopathology 2021, 79, 661–666. [Google Scholar] [CrossRef]
- Chernock, R.D.; Rivera, B.; Borrelli, N.; Hill, D.A.; Fahiminiya, S.; Shah, T.; Chong, A.S.; Aqil, B.; Mehrad, M.; Giordano, T.J.; et al. Poorly differentiated thyroid carcinoma of childhood and adolescence: A distinct entity characterized by DICER1 mutations. Mod. Pathol. 2020, 33, 1264–1274. [Google Scholar] [CrossRef]
- Rooper, L.M.; Bynum, J.P.; Miller, K.P.; Lin, M.T.; Gagan, J.; Thompson, L.D.R.; Bishop, J.A. Recurrent DICER1 Hotspot Mutations in Malignant Thyroid Gland Teratomas: Molecular Characterization and Proposal for a Separate Classification. Am. J. Surg. Pathol. 2020, 44, 826–833. [Google Scholar] [CrossRef]
- Nosé, V. DICER1 gene alterations in thyroid diseases. Cancer Cytopathol. 2020, 128, 688–689. [Google Scholar] [CrossRef]
- Durieux, E.; Descotes, F.; Mauduit, C.; Decaussin, M.; Guyetant, S.; Devouassoux-Shisheboran, M. The co-occurrence of an ovarian Sertoli-Leydig cell tumor with a thyroid carcinoma is highly suggestive of a DICER1 syndrome. Virchows Arch. 2016, 468, 631–636. [Google Scholar] [CrossRef]
- Capezzone, M.; Robenshtok, E.; Cantara, S.; Castagna, M.G. Familial non-medullary thyroid cancer: A critical review. J. Endocrinol. Investig. 2021, 44, 943–950. [Google Scholar] [CrossRef]
- LiVolsi, V.A.; Baraban, E.; Baloch, Z.W. Familial thyroid carcinoma: The road less traveled in thyroid pathology—An update. Diagn. Histopathol. 2017, 23, 366–377. [Google Scholar] [CrossRef]
- Park, J.-I.; Starenki, D. Pediatric Medullary Thyroid Carcinoma. J. Pediatr. Oncol. 2015, 3, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.D.; Mehrotra, S.; Kilic, A.I.; Toslak, I.E.; Lim-Dunham, J.; Martini, M.; Fadda, G.; Lombardi, C.P.; Larocca, L.M.; Barkan, G.A. Noninvasive follicular thyroid neo-plasm with papillary-like nuclear features in the pediatric age group. Cancer Cytopathol. 2018, 126, 27–35. [Google Scholar] [CrossRef]
- Massimino, M.; Evans, D.B.; Podda, M.; Spinelli, C.; Collini, P.; Pizzi, N.; Bleyer, A. Thyroid cancer in adolescents and young adults. Pediatr. Blood Cancer 2018, 65, e27025. [Google Scholar] [CrossRef]
- Collini, P.; Mattavelli, F.; Pellegrinelli, A.; Barisella, M.; Ferrari, A.; Massimino, M. Papillary carcinoma of the thyroid gland of childhood and adolescence: Morphologic subtypes, biologic behavior and prognosis: A clinicopathologic study of 42 sporadic cases treated at a single institution during a 30-year period. Am. J. Surg. Pathol. 2006, 30, 1420–1426. [Google Scholar] [CrossRef]
- Onder, S.; Ozturk Sari, S.; Yegen, G.; Sormaz, I.C.; Yilmaz, I.; Poyrazoglu, S.; Sanlı, Y.; Giles Senyurek, Y.; Kapran, Y.; Mete, O. Classic Architecture with Multicentricity and Local Recurrence, and Absence of TERT Promoter Mutations are Correlates of BRAF (V600E) Harboring Pediatric Papillary Thyroid Carcinomas. Endocr. Pathol. 2016, 27, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Cordioli, M.I.C.V.; Moraes, L.; Cury, A.N.; Cerutti, J.M. Are we really at the dawn of understanding sporadic pediatric thyroid carcinoma? Endocr. Relat. Cancer 2015, 22, R311–R324. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Mitsutake, N.; Saenko, V. Molecular pathogenesis of pediatric thyroid carcinoma. J. Radiat. Res. 2021, 62, i71–i77. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Kondo, T.; Nakazawa, T.; Mochizuki, K.; Inoue, T.; Kasai, K.; Tahara, I.; Yabuta, T.; Hirokawa, M.; Miyauchi, A.; et al. Frequent BRAF V600E and Absence of TERT Promoter Mutations Characterize Sporadic Pediatric Papillary Thyroid Carcinomas in Japan. Endocr. Pathol. 2017, 28, 103–111. [Google Scholar] [CrossRef]
- Henke, L.E.; Perkins, S.M.; Pfeifer, J.D.; Ma, C.; Chen, Y.; Dewees, T.; Grigsby, P.W. BRAF V600E mutational status in pediatric thyroid cancer. Pediatr. Blood Cancer 2014, 61, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Nikita, M.E.; Jiang, W.; Cheng, S.M.; Hantash, F.M.; McPhaul, M.J.; Newbury, R.O.; Phillips, S.A.; Reitz, R.E.; Waldman, F.M.; Newfield, R.S. Mutational analysis in pediatric thyroid cancer and correlations with age, ethnicity, and clinical presentation. Thyroid 2016, 26, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsutake, N.; Fukushima, T.; Matsuse, M.; Rogounovitch, T.; Saenko, V.; Uchino, S.; Ito, M.; Suzuki, K.; Suzuki, S.; Yamashita, S. BRAF(V600E) mutation is highly prevalent in thyroid carcinomas in the young population in Fukushima: A different oncogenic profile from Chernobyl. Sci. Rep. 2015, 5, 16976. [Google Scholar] [CrossRef] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Galuppini, F.; Vianello, F.; Censi, S.; Barollo, S.; Bertazza, L.; Carducci, S.; Colato, C.; Manso, J.; Rugge, M.; Iacobone, M.; et al. Differentiated Thyroid Carcinoma in Pediatric Age: Genetic and Clinical Scenario. Front. Endocrinol. 2019, 10, 552. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Shakya, S.; Ballal, S.; Agarwal, S.; Bal, C. BRAF V600E and TERT promoter mutations in paediatric and young adult papillary thyroid cancer and clinicopathological correlation. J. Pediatr. Endocrinol. Metab. 2020, 33, 1465–1474. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Murugan, A.K.; Qasem, E.; Alswailem, M.; Al-Hindi, H.; Shi, Y. Single Point Mutations in Pediatric Differentiated Thyroid Cancer. Thyroid 2017, 27, 189–196. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Sisdelli, L.; Cordioli, M.I.V.; Vaisman, F.; Caria, P.; Mai, S.; Cerutti, J.M. Genetic landscape of papillary thyroid carcinoma and nuclear architecture: An overview comparing pediatric and adult populations. Cancers 2020, 12, 3146. [Google Scholar] [CrossRef]
- Cordioli, M.I.C.V.; Moraes, L.; Carvalheira, G.; Sisdelli, L.; Alves, M.T.S.; Delcelo, R.; Monte, O.; Longui, C.A.; Cury, A.N.; Cerutti, J.M. AGK-BRAF gene fusion is a recurrent event in sporadic pediatric thyroid carcinoma. Cancer Med. 2016, 5, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Sisdelli, L.; Cordioli, M.I.C.V.; Vaisman, F.; Moraes, L.; Colozza-Gama, G.A.; Alves, P.A.G.; Araújo, M.L.; Alves, M.T.S.; Monte, O.; Longui, C.A.; et al. AGK-BRAF is associated with distant metastasis and younger age in pediatric papillary thyroid carcinoma. Pediatr. Blood Cancer 2019, 66, e27707. [Google Scholar] [CrossRef]
- Cordioli, M.I.C.V.; Moraes, L.; Bastos, A.U.; Besson, P.; Alves, M.T.D.S.; Delcelo, R.; Monte, O.; Longui, C.; Cury, A.N.; Cerutti, J.M. Fusion Oncogenes Are the Main Genetic Events Found in Sporadic Papillary Thyroid Carcinomas from Children. Thyroid 2017, 27, 182–188. [Google Scholar] [CrossRef]
- Pekova, B.; Sykorova, V.; Dvorakova, S.; Vaclavikova, E.; Moravcova, J.; Katra, R.; Astl, J.; Vlcek, P.; Kodetova, D.; Vcelak, J.; et al. RET, NTRK, ALK, BRAF, and MET Fusions in a Large Cohort of Pediatric Papillary Thyroid Carcinomas. Thyroid 2020, 30, 1771–1780. [Google Scholar] [CrossRef]
- Efanov, A.A.; Brenner, A.V.; Bogdanova, T.I.; Kelly, L.M.; Liu, P.; Little, M.P.; Wald, A.I.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. Investigation of the Relationship Between Radiation Dose and Gene Mutations and Fusions in Post-Chernobyl Thyroid Cancer. J. Natl. Cancer Inst. 2018, 110, 371–378. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Hosler, G.; Zahurak, M.; Cohen, Y.; Sidransky, D.; Westra, W.H. Mutational activation of BRAF is not a major event in sporadic childhood papillary thyroid carcinoma. Mod. Pathol. 2005, 18, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Marotta, V.; Bifulco, M.; Vitale, M. Significance of RAS Mutations in Thyroid Benign Nodules and Non-Medullary Thyroid Cancer. Cancers 2021, 13, 3785. [Google Scholar] [CrossRef]
- Kumagai, A.; Namba, H.; Saenko, V.A.; Ashizawa, K.; Ohtsuru, A.; Ito, M.; Ishikawa, N.; Sugino, K.; Ito, K.; Jeremiah, S.; et al. Low frequency of BRAFT1796A mutations in childhood thyroid carcinomas. J. Clin. Endocrinol. Metab. 2004, 89, 4280–4284. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.T.; Labourier, E.; Ablordeppey, K.K.; Surrey, L.F.; Mostoufi-Moab, S.; Isaza, A.; Adzick, N.S.; Kazahaya, K.; Kumar, G.; Bauer, A.J. miRNA expression can classify pediatric thyroid lesions and increases the diagnostic yield of mutation testing. Pediatr. Blood Cancer 2020, 67, e28276. [Google Scholar] [CrossRef]
- Lee, Y.A.; Lee, H.; Im, S.W.; Song, Y.S.; Oh, D.Y.; Kang, H.J.; Won, J.K.; Jung, K.C.; Kwon, D.; Chung, E.J.; et al. NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J. Clin. Investig. 2021, 131, e144847. [Google Scholar] [CrossRef]
- Geng, J.; Liu, Y.; Guo, Y.; Wang, H.; Tai, J.; Jin, Y.; Zhang, J.; Yu, Y.; Wang, S.; Song, Y.; et al. Correlation between TERT C228T and clinic-pathological features in pediatric papillary thyroid carcinoma. Sci. China Life Sci. 2019, 62, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Vyas, M.; Horne, M.J.; Virk, R.K.; Morotti, R.; Liu, Z.; Tallini, G.; Nikiforova, M.N.; Christison-Lagay, E.R.; Udelsman, R.; et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer 2016, 122, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Propst, E.J.; Wasserman, J.D.; Gorodensky, J.; Ngan, B.Y.; Wolter, N.E. Patterns and Predictors of Metastatic Spread to the Neck in Pediatric Thyroid Carcinoma. Laryngoscope 2021, 131, E1002–E1009. [Google Scholar] [CrossRef] [PubMed]
- Aschebrook-Kilfoy, B.; Grogan, R.H.; Ward, M.H.; Kaplan, E.; Devesa, S.S. Follicular Thyroid Cancer Incidence Patterns in the United States, 1980–2009. Thyroid 2013, 23, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.G.; Kondo, T.; Oishi, N.; Nakazawa, T.; Mochizuki, K.; Miyauchi, A.; Hirokawa, M.; Katoh, R. Paediatric follicular thyroid carcinoma—Indolent cancer with low prevalence of RAS mutations and absence of PAX8–PPARG fusion in a Japanese population. Histopathology 2017, 71, 760–768. [Google Scholar] [CrossRef]
- Ito, Y.; Miyauchi, A.; Tomoda, C.; Hirokawa, M.; Kobayashi, K.; Miya, A. Prognostic significance of patient age in minimally and widely invasive follicular thyroid carcinoma: Investigation of three age groups. Endocr. J. 2014, 61, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Fukahori, M.; Yoshida, A.; Hayashi, H.; Yoshihara, M.; Matsukuma, S.; Sakuma, Y.; Koizume, S.; Okamoto, N.; Kondo, T.; Masuda, M.; et al. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: New insights from a single center and a large patient cohort. Thyroid 2012, 22, 683–689. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Biddinger, P.W.; Caudill, C.M.; Kroll, T.G.; Nikiforov, Y.E. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am. J. Surg. Pathol. 2002, 26, 1016–1023. [Google Scholar] [CrossRef]
- Yoo, S.K.; Lee, S.; Kim, S.J.; Jee, H.G.; Kim, B.A.; Cho, H.; Song, Y.S.; Cho, S.W.; Won, J.K.; Shin, J.Y.; et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet. 2016, 12, e1006239. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- Mostoufi-Moab, S.; Labourier, E.; Sullivan, L.; Livolsi, V.; Li, Y.; Xiao, R.; Beaudenon-Huibregtse, S.; Kazahaya, K.; Scott Adzick, N.; Baloch, Z.; et al. Molecular Testing for Oncogenic Gene Alterations in Pediatric Thyroid Lesions. Thyroid 2018, 28, 60–67. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Sarabia, S.F.; Sayeed, H.; Patel, N.; Baalwa, J.; Athanassaki, I.; Hernandez, J.A.; Fang, E.; Quintanilla, N.M.; Roy, A.; et al. Integrating molecular testing in the diagnosis and management of children with thyroid lesions. Pediatr. Dev. Pathol. 2016, 19, 94–100. [Google Scholar] [CrossRef]
- Suster, D.; Michal, M.; Nishino, M.; Piana, S.; Bongiovanni, M.; Blatnik, O.; Hájková, V.; Ptáková, N.; Michal, M.; Suster, S. Papillary thyroid carcinoma with prominent myofibroblastic stromal component: Clinicopathologic, immunohistochemical and next-generation sequencing study of seven cases. Mod. Pathol. 2020, 33, 1702–1711. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front. Endocrinol. 2012, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Sastre-Perona, A.; Riesco-Eizaguirre, G.; Zaballos, M.A.; Santisteban, P. β-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget 2016, 7, 49435–49449. [Google Scholar] [CrossRef] [Green Version]
- Kremenevskaja, N.; Von Wasielewski, R.; Rao, A.S.; Schöfl, C.; Andersson, T.; Brabant, G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar] [CrossRef] [Green Version]
- Ngeow, J.; Mester, J.; Rybicki, L.A.; Ni, Y.; Milas, M.; Eng, C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J. Clin. Endocrinol. Metab. 2011, 96, E2063–E2071. [Google Scholar] [CrossRef]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Rivkees, S.A.; Mazzaferri, E.L.; Verburg, F.A.; Reiners, C.; Luster, M.; Breuer, C.K.; Dinauer, C.A.; Udelsman, R. The Treatment of Differentiated Thyroid Cancer in Children: Emphasis on Surgical Approach and Radioactive Iodine Therapy. Endocr. Rev. 2011, 32, 798–826. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, M.L.; Schmidt, A.; Al Ghuzlan, A.; Lacroix, L.; de Vathaire, F.; Chevillard, S.; Schlumberger, M. Radiation exposure and thyroid cancer: A review. Arch. Endocrinol. Metab. 2017, 61, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Yasui, Y.; Robison, L.L.; Birch, J.M.; Bogue, M.K.; Diller, L.; DeLaat, C.; Fossati-Bellani, F.; Morgan, E.; Oberlin, O.; et al. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: Report from the Late Effects Study Group. J. Clin. Oncol. 2003, 21, 4386–4394. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.Z.; Cibas, E.S. (Eds.) The Bethesda System for Reporting Thyroid Cytopathology: Definitions, Criteria, and Explanatory Notes, 2nd ed.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. (Eds.) Chapter 2—Tumours of the Thyroid Gland. In WHO Classification of Tumours of Endocrine Organ; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 65–143. [Google Scholar]
- Livolsi, V.A. Papillary thyroid carcinoma: An update. Mod. Pathol. 2011, 24, S1–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.S.; Hong, S.; Park, C.S. Diffuse Sclerosing Variant Is a Major Subtype of Papillary Thyroid Carcinoma in the Young. Thyroid 2009, 19, 1225–1231. [Google Scholar] [CrossRef]
- Canberk, S.; Ferreira, J.C.; Pereira, L.; Batlsta, R.; Vieira, A.F.; Soares, P.; Sobrinho Simões, M.; Máximo, V. Analyzing the Role of DICER1 Germline Variations in Papillary Thyroid Carcinoma. Eur. Thyroid J. 2021, 9, 296–303. [Google Scholar] [CrossRef]
- Motomura, T.; Nikiforov, Y.E.; Namba, H.; Ashizawa, K.; Nagataki, S.; Yamashita, S.; Fagin, J.A. ret rearrangements in Japanese pediatric and adult papillary thyroid cancers. Thyroid 1998, 8, 485–489. [Google Scholar] [CrossRef]
- Geng, J.; Wang, H.; Liu, Y.; Tai, J.; Jin, Y.; Zhang, J.; He, L.; Fu, L.; Qin, H.; Song, Y.; et al. Correlation between BRAF V600E mutation and clinicopathological features in pediatric papillary thyroid carcinoma. Sci. China Life Sci. 2017, 60, 729–738. [Google Scholar] [CrossRef]
- Kure, S.; Ishino, K.; Kudo, M.; Wada, R.; Saito, M.; Nagaoka, R.; Sugitani, I.; Naito, Z. Incidence of BRAF V600E mutation in patients with papillary thyroid carcinoma: A single-institution experience. J. Int. Med. Res. 2019, 47, 5560–5572. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, M.; Mitsutake, N.; Matsuse, M.; Fukushima, T.; Suzuki, S.; Matsumoto, Y.; Ookouchi, C.; Mizunuma, H.; Nakamura, I.; Nakano, K.; et al. The Clinicopathological Results of Thyroid Cancer with BRAFV600E Mutation in the Young Population of Fukushima. J. Clin. Endocrinol. Metab. 2020, 105, e4328–e4336. [Google Scholar] [CrossRef]
- Zhao, Z.; Yin, X.D.; Zhang, X.H.; Li, Z.W.; Wang, D.W. Comparison of pediatric and adult medullary thyroid carcinoma based on SEER program. Sci. Rep. 2020, 10, 13310. [Google Scholar] [CrossRef]
- Moline, J.; Eng, C. Multiple endocrine neoplasia type 2: An overview. Genet. Med. 2011, 13, 755–764. [Google Scholar] [CrossRef]
- Carlson, K.M.; Dou, S.; Chi, D.; Scavarda, N.; Toshima, K.; Jackson, C.E.; Wells, S.A.; Goodfellow, P.J.; Donis-Keller, H. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc. Natl. Acad. Sci. USA 1994, 91, 1579–1583. [Google Scholar] [CrossRef]
- Wells, S.A.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; MacHens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Labourier, E.; Shifrin, A.; Busseniers, A.E.; Lupo, M.A.; Manganelli, M.L.; Andruss, B.; Wylie, D.; Beaudenon-Huibregtse, S. Molecular Testing for miRNA, mRNA, and DNA on Fine-Needle Aspiration Improves the Preoperative Diagnosis of Thyroid Nodules with Indeterminate Cytology. J. Clin. Endocrinol. Metab. 2015, 100, 2743–2750. [Google Scholar] [CrossRef]
- Durante, C.; Puxeddu, E.; Ferretti, E.; Morisi, R.; Moretti, S.; Bruno, R.; Barbi, F.; Avenia, N.; Scipioni, A.; Verrienti, A.; et al. BRAF Mutations in Papillary Thyroid Carcinomas Inhibit Genes Involved in Iodine Metabolism. J. Clin. Endocrinol. Metab. 2007, 92, 2840–2843. [Google Scholar] [CrossRef] [Green Version]
- Cordioli, M.I.C.V.; Moraes, L.; Alves, M.T.D.S.; Delcelo, R.; Monte, O.; Longui, C.A.; Cury, A.N.; Cerutti, J.M. Thyroid-specific genes expression uncovered age-related differences in pediatric thyroid carcinomas. Int. J. Endocrinol. 2016, 2016, 1956740. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.A.; Fahey, T.J. Molecular markers in thyroid cancer diagnostics. Surg. Clin. N. Am. 2009, 89, 1139–1155. [Google Scholar] [CrossRef] [PubMed]
- Al Ghuzlan, A.; Caillou, B.; Schlumberger, M. Galectin-3 for indeterminate thyroid cytology. Lancet Oncol. 2008, 9, 508–510. [Google Scholar] [CrossRef] [PubMed]
- de Matos, L.L.; Del Giglio, A.B.; Matsubayashi, C.O.; de Lima Farah, M.; Del Giglio, A.; da Silva Pinhal, M.A. Expression of CK-19, galectin-3 and HBME-1 in the differentiation of thyroid lesions: Systematic review and diagnostic meta-analysis. Diagn. Pathol. 2012, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Prasad, P.K.; Mahajan, P.; Hawkins, D.S.; Mostoufi-Moab, S.; Venkatramani, R. Management of pediatric differentiated thyroid cancer: An overview for the pediatric oncologist. Pediatr. Blood Cancer 2020, 67, e28141. [Google Scholar] [CrossRef]
- Casado-Medrano, V.; O’Neill, A.; Halada, S.; Laetsch, T.W.; Bauer, A.J.; Franco, A.T. NTRK-fusions in pediatric thyroid tumors: Current state and future perspectives. Cancer Genet. 2022, 264–265, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mehrad, M.; Ely, K.A.; Liang, J.; Solórzano, C.C.; Neblett, W.W., III; Coogan, A.C.; Weiss, V.L. Incidence and malignancy rates of indeterminate pediatric thyroid nodules. Cancer Cytopathol. 2019, 127, 231–239. [Google Scholar] [CrossRef]
- Kim, M.I.; Alexander, E.K. Diagnostic use of molecular markers in the evaluation of thyroid nodules. Endocr. Pract. 2012, 18, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Vella, C.; Baldacchino, S.; Formosa, R.; Vassallo, J. The Utility of Galectin-3 and HBME-1 Immunohistochemical Expression in Thyroid Cancer in the Maltese Population. Endocrines 2022, 3, 225–239. [Google Scholar] [CrossRef]
- Buryk, M.A.; Monaco, S.E.; Witchel, S.F.; Mehta, D.K.; Gurtunca, N.; Nikiforov, Y.E.; Simons, J.P. Preoperative cytology with molecular analysis to help guide surgery for pediatric thyroid nodules. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1697–1700. [Google Scholar] [CrossRef]
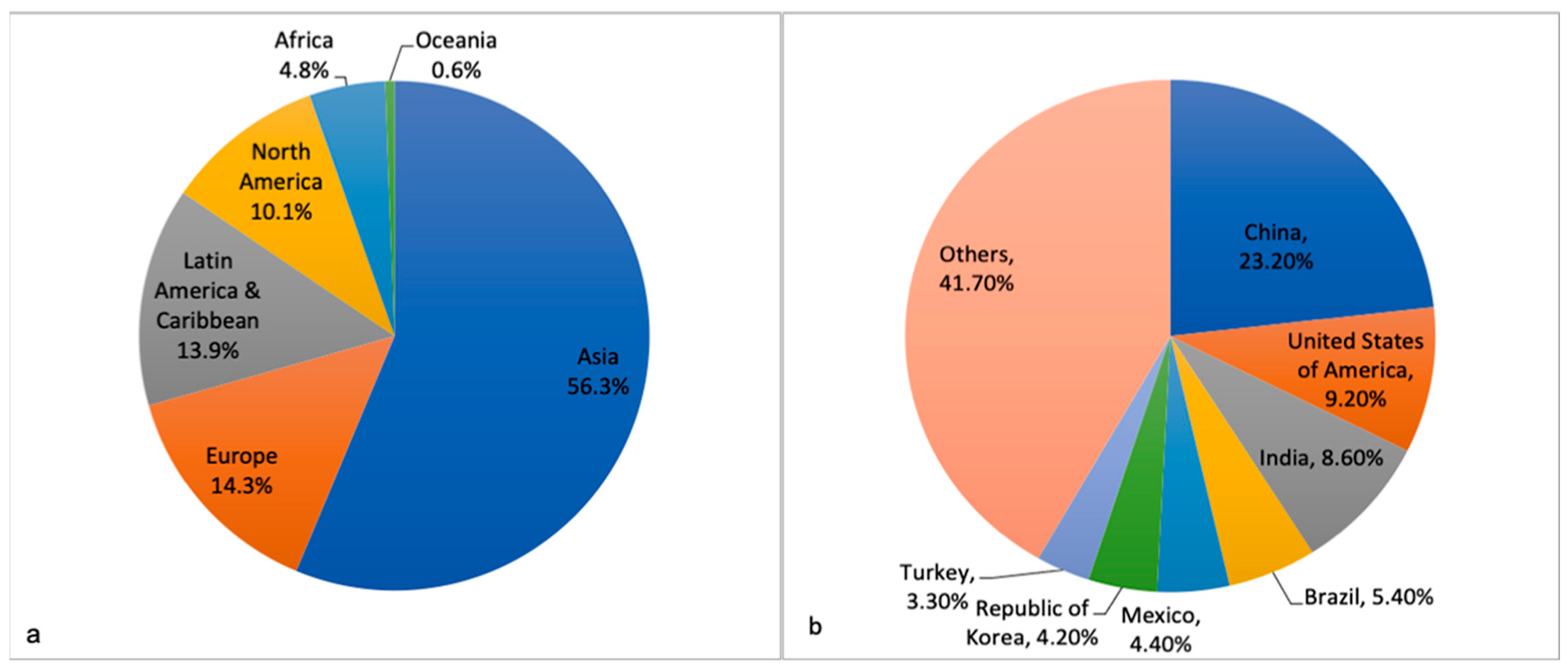

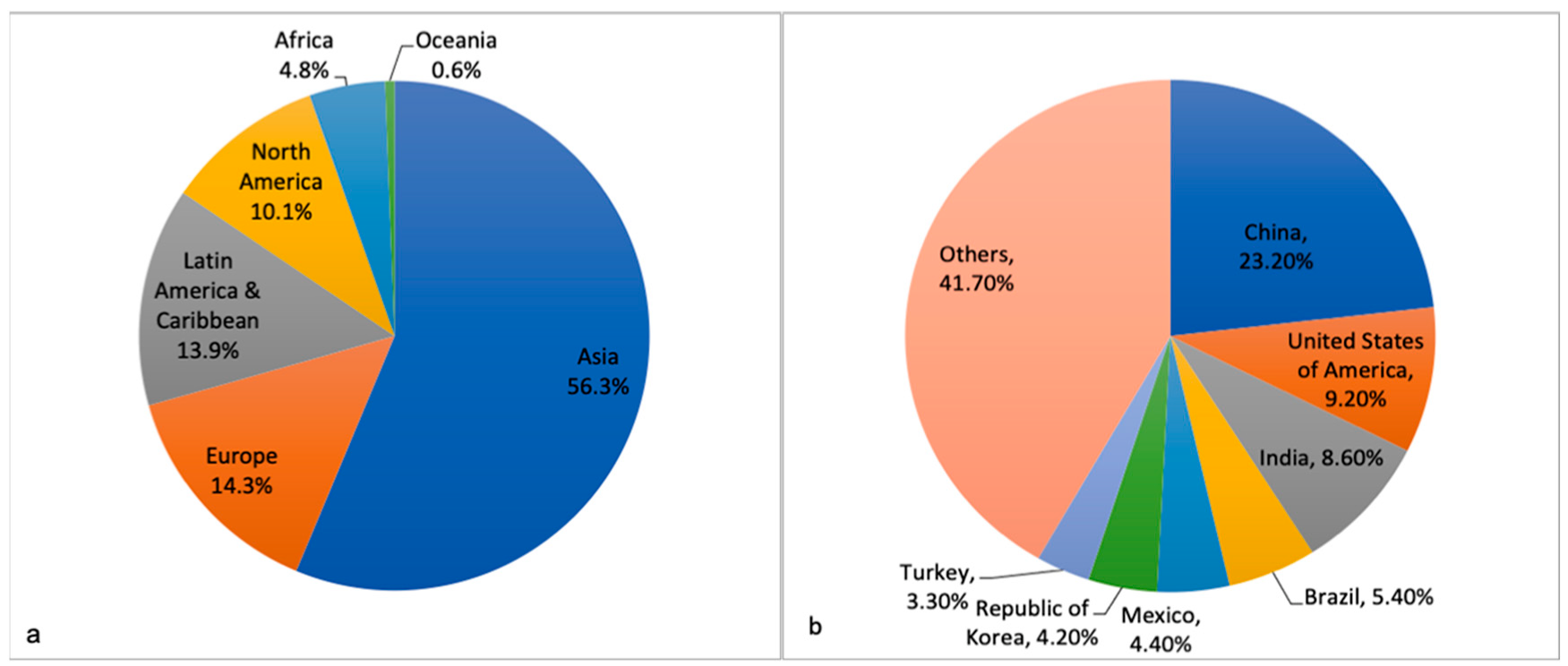{kind=link}
| Characteristics | Adults | References | Pediatric Age Group | References |
|---|---|---|---|---|
| Clinical symptoms | Hoarseness, dysphagia, cough. Nodule common but rarely (<5–10%) malignant | [14,59] | Asymptomatic, incidentally detected. Nodule uncommon but frequently (22–26%) malignant if present | [14,59] |
| Upfront lymph node involvement | 20–50% | [103] | 40–90% | [103] |
| Distant metastasis | 2% | [103] | 20–30% | [103] |
| Radiation exposure | Risk of cancer less | [104] | 5–10 fold increased risk; higher rates reported post-radiotherapy | [104,105] |
FNAC
| Not mandatory <1cm: FNAC usually not recommended FNAC not recommended Recommended 6–18% Repeat FNAC/molecular testing/lobectomy 10–40% Molecular testing/lobectomy Recommended | [6] [6,103] [6] [6,15] [14,106] [106] [14,106] [106] [69] | Mandatory Size not a criterion for decision making FNAC not recommended (should be removed surgically) Recommended 28% Lobectomy plus isthmusectomy 58% Lobectomy plus isthmusectomy Not recommended | [6] [6,103] [6] [6,15] [6,14,15] [6] [14] [6] [6,69] |
| Tumor classification system | AJCC TNM classification | [6] | AJCC TNM classification with ATA risk-stratification system * | [6] |
| 5-year relative survival | 98.3% | [3] | 99.7% | [3] |
| PTC multifocality | 20% | [107] | 65% | [6] |
| Histopathological subtypes of PTC | High risk subtypes less common (<20%) | [108] | Classic PTC 20–50%; high risk subtypes (tall cell, diffuse sclerosing, solid/trabecular) form 15–40% | [14,60,61,109] |
| Molecular profile (PTC) BRAF V600E | 30–90% | [107] | 0–63% (sporadic) 0–70% (post-radiation) | [14,73] |
| BRAF fusions | <3% | [63,73] | 0–20% (sporadic) 0–11% (post-radiation) | [14,73] |
| RET fusions | 5–35% | [107] | 22–65% (sporadic) 33–77% (post-radiation) | [62,73] |
| H-/K-/N-RAS mutations | 0–35% | [107] | <10% | [1,72] |
| TERT promoter mutations (C250T, C228T) | 5–25% | [107] | 0–27% | [73,83] |
| NTRK fusions | 1–5% | [73] | 0–20% (sporadic) 1–15% (post-radiation) | [73,77] |
| PAX8/PPARG fusion | 0–5% | [73] | 0–9% (sporadic) 4% (post-radiation) | [73] |
| DICER1 mutations | ∼4% ** | [110] | 7.6% | [83] |
| ALK fusions | 0–3% | [73] | 0–7% (sporadic) 1–7% (post-radiation) | [73] |
| Molecular profile (FTC) H-/K-/N-RAS mutations | 10–57% | [90,91,93,94] | 0–50% | [14,82,88] |
| PAX8/PPARG fusion | 35–50% | [90,92,93,94] | 0–20% | [67,88,95] |
| PTEN mutations | <1% | [63] | <2% (sporadic) 25% (carriers of PTEN mutation) | [72,101] |
| DICER1 mutations | 1% | [47] | 25–53% *** | [47,49] |
| Studies | Year | Country | Age Range (Years) | n | Sporadic/Post Radiation | Subtypes | Molecular Method Used | BRAF V600E | RET/PTC | BRAF Fusion | TERT Promoter Mutation | RAS Mutation | NTRK Fusions | Additional Alterations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Motomura et al. [111] | 1998 | Japan | 9–14 | 10 | Sporadic | Classical (6) F-PTC (2) DS-PTC (1) S-PTC (1) | RT-PCR followed by southern hybridization | - | RET/PTC1 20% RET/PTC3 10% | - | - | - | - | - |
| Kumagai et al. [81] | 2005 | Japan | <15 | 31 | Sporadic | Classical (27) F-PTC (2) FTC (1) PDTC (1) | Direct sequencing, RT-PCR | 3.2% | - | - | - | 0 | - | - |
| ≤15 | 15 | Radiation # | Classical (2) S-PTC (4) F-PTC (2) Mixed (7) | 0 | - | - | - | 0 | 33.3% | - | ||||
| 15–31 | 33 | Radiation # | Classical (7) S-PTC (1) F-PTC (9) Mixed (16) | 24.2% | - | - | - | 6.1% | 36.4% | - | ||||
| Mitsutake et al. [68] | 2015 | Japan | ≤22 | 68 | Likely sporadic $ | Classical (61) F-PTC (2) CMTC (4) PDTC (1) | Direct sequencing, RT-PCR | 63.2% | RET/PTC1 8.8% RET/PTC3 1.5% | 0 | 0 | 0 | 5.9% | - |
| Alzahrani et al. [7] | 2016 | Saudi Arabia | ≤18 | 55 | Sporadic | Classical (44) FV (6) TC-PTC (1) DS-PTC (1) FTC (2) PDTC (1) | Direct sequencing | 22.6% | - | - | 1.8% | - | - | - |
| Alzahrani et al. * [72] | 2017 | Saudi Arabia | ≤18 yrs | 79 | Sporadic | Classical (72) F-PTC (7) | Direct sequencing | 24.1% | - | - | 1.3% | 2.5% | - | PIK3CA exon 9: 1.4% PIK3CA exon 20: 1.3% PTEN exon 5: 1.4% |
| Geng et al. ° [112] | 2017 | China | 3–13 | 48 | Sporadic | Classical (41) F-PTC (5) DS-PTC (2) | Direct sequencing | 35.4% | - | - | - | - | - | - |
| Oishi et al. [65] | 2017 | Japan | ≤20 | 81 | Sporadic | Classical (66) CMTC (1) F-PTC (2) DS-PTC (4) S-PTC (8) | Allele specific PCR and/or Sanger sequencing | 54% | - | - | 0% | - | - | - |
| Vuong et al. [88] | 2017 | Japan | <21 | 41 | Sporadic | FTC (41) | Direct sequencing and RT-PCR | - | - | - | - | 12.2% | - | 0% (PAX8/PPARG) |
| Geng et al. ° [84] | 2019 | China | 3–13 | 48 | Sporadic | Classical (41) F-PTC (5) DS-PTC (2) | Direct sequencing | - | - | - | 27.1% | - | - | - |
| Kure et al. [113] | 2019 | Japan | 13–19 | 7 | Sporadic | Classical (5) CMTC (1) Classical with poorly differentiated component (1) | Direct sequencing | 29% | - | - | - | - | - | - |
| Iwadate el al. & [114] | 2020 | Japan | 0–24 | 138 | Likely sporadic $ | Classical (125) CMTC (4) F-PTC (3) DS-PTC (2) S-PTC (2) PDTC (1) Others (1) | Direct sequencing and RT-PCR | 70.6% of PTCs | RET/PTC1 5.9% of PTCs RET/PTC3 0.7% of PTCs | - | - | - | 5.9% of PTCs | AFAP1L2/RET (0.7% of PTCs) PPFIBP2/RET (0.7% of PTCs) STRN/ALK (1.5% of PTCs) KIAA1217/RET (0.7% of PTCs) Delta RFP/RET (0.7% of PTCs) |
| Chakraborty et al. [71] | 2020 | India | ≤20 | 100 | Sporadic | Classical (72) F-PTC (24) TC-PTC (2) FTC (2) | Direct sequencing | 14% | - | - | 0% | - | - | - |
| Lee et al. [49] | 2020 | Republic of Korea | <20 | 15 | 10 sporadic, 4 DICER1 syndrome, 1 PTEN hamartoma syndrome | FTC | WES, targeted NGS, direct sequencing | - | - | - | 0% | 0% | - | DICER1 (53.3%), PTEN (6.7%), PAX8/PPARG (6.7%) |
| Bae et al. [47] | 2021 | Japan, Republic of Korea | <18 | 41 | Sporadic | Follicular-patterned tumors ∗ | Targeted NGS | 0% | 0% | 0% | 0% | 20% | 0% | DICER1 (22%), FGFR3 (15%), PTEN (12%), STK11 (10%), APC (5%), TSHR (5%), CTNNB1 (2%), TP53 (2%), EIF1AX (2%), FGFR4 (2%), GNAS (2%), RET (2%), SOS1 (2%), THADA/IGF2BP3 (2%) |
| Lee et al. [83] | 2021 | Republic of Korea | <10 | 14 | Sporadic and radiation ^ | Classical (75) DS-PTC (14) Others (15) | NGS, direct sequencing, FISH, and/or IHC. | 0% | 64.3% | - | 7.1% | 0% | 14.3% | 0% (DICER1) |
| 10–15 | 40 | 27.5% | 20% | - | 0% | 0% | 5% | 2.5% (DICER1) | ||||||
| 15–20 | 52 | 57.7% | 7.7% | - | 1.9% | 0% | 0% | 7.7% (DICER1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guleria, P.; Srinivasan, R.; Rana, C.; Agarwal, S. Molecular Landscape of Pediatric Thyroid Cancer: A Review. Diagnostics 2022, 12, 3136. https://doi.org/10.3390/diagnostics12123136
Guleria P, Srinivasan R, Rana C, Agarwal S. Molecular Landscape of Pediatric Thyroid Cancer: A Review. Diagnostics. 2022; 12(12):3136. https://doi.org/10.3390/diagnostics12123136
Chicago/Turabian StyleGuleria, Prerna, Radhika Srinivasan, Chanchal Rana, and Shipra Agarwal. 2022. "Molecular Landscape of Pediatric Thyroid Cancer: A Review" Diagnostics 12, no. 12: 3136. https://doi.org/10.3390/diagnostics12123136
APA StyleGuleria, P., Srinivasan, R., Rana, C., & Agarwal, S. (2022). Molecular Landscape of Pediatric Thyroid Cancer: A Review. Diagnostics, 12(12), 3136. https://doi.org/10.3390/diagnostics12123136