
CSF Diagnostics: A Potentially Valuable Tool in Neurodegenerative and Inflammatory Disorders Involving Motor Neurons: A Review


 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Amyotrophic Lateral Sclerosis
3.1. Cytokines
3.2. TDP-43
3.3. Neurofilaments
4. Peripheral Neuropathies
4.1. Guillain-Barré Syndrome
4.1.1. Neurofilaments
4.1.2. Sphingomyelin and Lipidomics
4.1.3. Proteins
Cystatin C
Transthyretin
Haptoglobin
Tau
Cytokines
5. Multifocal Motor Neuropathy
6. Neuroborreliosis
7. Spinal Muscular Atrophy
7.1. Routine CSF Parameters in SMA
7.2. Protein Biomarkers in SMA
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Benninger, F.; Steiner, I. CSF in acute and chronic infectious diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 146, pp. 187–206. [Google Scholar] [CrossRef]
- Studahl, M.; Lindquist, L.; Eriksson, B.-M.; Günther, G.; Bengner, M.; Franzen-Röhl, E.; Fohlman, J.; Bergström, T.; Aurelius, E. Acute Viral Infections of the Central Nervous System in Immunocompetent Adults: Diagnosis and Management. Drugs 2013, 73, 131–158. [Google Scholar] [CrossRef]
- Shalaby, T.; Grotzer, M.A. Tumor-Associated CSF MicroRNAs for the Prediction and Evaluation of CNS Malignancies. Int. J. Mol. Sci. 2015, 16, 29103–29119. [Google Scholar] [CrossRef]
- McEwen, A.E.; Leary, S.E.S.; Lockwood, C.M. Beyond the Blood: CSF-Derived cfDNA for Diagnosis and Characterization of CNS Tumors. Front. Cell Dev. Biol. 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H.; Minthon, L.; Lannfelt, L.; Strid, S.; Annas, P.; Basun, H.; Andreasen, N. Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci. Lett. 2007, 419, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Buchhave, P.; Blennow, K.; Zetterberg, H.; Stomrud, E.; Londos, E.; Andreasen, N.; Minthon, L.; Hansson, O. Longitudinal Study of CSF Biomarkers in Patients with Alzheimer’s Disease. PLoS ONE 2009, 4, e6294. [Google Scholar] [CrossRef] [PubMed]
- Brinkmalm, G.; Sjödin, S.; Simonsen, A.H.; Hasselbalch, S.G.; Zetterberg, H.; Brinkmalm, A.; Blennow, K. A Parallel Reaction Monitoring Mass Spectrometric Method for Analysis of Potential CSF Biomarkers for Alzheimer’s Disease. Proteom. Clin. Appl. 2017, 12. [Google Scholar] [CrossRef]
- Anoop, A.; Singh, P.K.; Jacob, R.S.; Maji, S.K. CSF Biomarkers for Alzheimer’s Disease Diagnosis. Int. J. Alzheimer’s Dis. 2010, 2010, 606802. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Farotti, L.; Paciotti, S.; Tasegian, A.; Eusebi, P.; Parnetti, L. Discovery, validation and optimization of cerebrospinal fluid biomarkers for use in Parkinson’s disease. Expert Rev. Mol. Diagn. 2017, 17, 771–780. [Google Scholar] [CrossRef]
- Goldman, J.G.; Andrews, H.; Amara, A.; Naito, A.; Alcalay, R.N.; Shaw, L.M.; Taylor, P.; Xie, T.; Tuite, P.; Henchcliffe, C.; et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: Relationships among biomarkers and Parkinson’s disease Features. Mov. Disord. 2017, 33, 282–288. [Google Scholar] [CrossRef]
- Katayama, T.; Sawada, J.; Takahashi, K.; Yahara, O. Cerebrospinal Fluid Biomarkers in Parkinson’s Disease: A Critical Overview of the Literature and Meta-Analyses. Brain Sci. 2020, 10, 466. [Google Scholar] [CrossRef]
- Mollenhauer, B.; Caspell-Garcia, C.J.; Coffey, C.S.; Taylor, P.; Shaw, L.M.; Trojanowski, J.Q.; Singleton, A.; Frasier, M.; Marek, K.; Galasko, D.; et al. Longitudinal CSF biomarkers in patients with early Parkinson disease and healthy controls. Neurology 2017, 89, 1959–1969. [Google Scholar] [CrossRef]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- Novakova, L.; Axelsson, M.; Khademi, M.; Zetterberg, H.; Blennow, K.; Malmeström, C.; Piehl, F.; Olsson, T.; Lycke, J. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J. Neurochem. 2016, 141, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Lycke, J.; Zetterberg, H. The role of blood and CSF biomarkers in the evaluation of new treatments against multiple sclerosis. Expert Rev. Clin. Immunol. 2017, 13, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Pietroboni, A.M.; Di Cola, F.S.; Scarioni, M.; Fenoglio, C.; Spanò, B.; Arighi, A.; Cioffi, S.M.; Oldoni, E.; A De Riz, M.; Basilico, P.; et al. CSF β-amyloid as a putative biomarker of disease progression in multiple sclerosis. Mult. Scler. J. 2016, 23, 1085–1091. [Google Scholar] [CrossRef]
- Masvekar, R.; Wu, T.; Kosa, P.; Barbour, C.; Fossati, V.; Bielekova, B. Cerebrospinal fluid biomarkers link toxic astrogliosis and microglial activation to multiple sclerosis severity. Mult. Scler. Relat. Disord. 2018, 28, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.R.; Søndergaard, H.B.; Buhelt, S.; von Essen, M.R.; Christensen, J.R.; Enevold, C.; Sellebjerg, F. Multiplex assessment of cerebrospinal fluid biomarkers in multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 45, 102391. [Google Scholar] [CrossRef]
- Magliozzi, R.; Cross, A.H. Can CSF biomarkers predict future MS disease activity and severity? Mult. Scler. J. 2020, 26, 582–590. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2017, 8, a024125. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.; Eisen, A.; Hardiman, O.; Burrell, J.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Hobson, E.V.; McDermott, C. Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2016, 12, 526–538. [Google Scholar] [CrossRef]
- Brooks, B.R. El escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 124, 96–107. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Ropper, A.H.; Samuels, M.A.; Klein, J.P.; Prasad, S. Adams and Victor’s Principles of Neurology, 11th ed.; McGraw-Hill: New York, NY, USA, 2019. [Google Scholar]
- Brodovitch, A.; Boucraut, J.; Delmont, E.; Parlanti, A.; Grapperon, A.-M.; Attarian, S.; Verschueren, A. Combination of serum and CSF neurofilament-light and neuroinflammatory biomarkers to evaluate ALS. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Oeckl, P.; Jardel, C.; Salachas, F.; Lamari, F.; Andersen, P.M.; Bowser, R.; De Carvalho, M.; Costa, J.; Van Damme, P.; Gray, E.; et al. Multicenter validation of CSF neurofilaments as diagnostic biomarkers for ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 404–413. [Google Scholar] [CrossRef]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Matsui, N.; Fujita, K.; Miyashiro, A.; Nodera, H.; Izumi, Y.; Shimizu, F.; Miyamoto, K.; Takahashi, Y.; Kanda, T.; et al. Increased proinflammatory cytokines in sera of patients with multifocal motor neuropathy. J. Neurol. Sci. 2014, 346, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Noto, Y.-I.; Shibuya, K.; Sato, Y.; Kanai, K.; Misawa, S.; Sawai, S.; Mori, M.; Uchiyama, T.; Isose, S.; Nasu, S.; et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph. Lateral Scler. 2010, 12, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.A.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2008, 117, 55–62. [Google Scholar] [CrossRef]
- Hosokawa, M.; Arai, T.; Yamashita, M.; Tsuji, H.; Nonaka, T.; Masuda-Suzukake, M.; Tamaoka, A.; Hasegawa, M.; Akiyama, H. Differential diagnosis of amyotrophic lateral sclerosis from Guillain–Barré syndrome by quantitative determination of TDP-43 in cerebrospinal fluid. Int. J. Neurosci. 2013, 124, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.F.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.-Y.; et al. TDP-43 in Cerebrospinal Fluid of Patients With Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; Arnim, C.A.F.V.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2015, 87, 12–20. [Google Scholar] [CrossRef]
- Tateishi, T.; Yamasaki, R.; Tanaka, M.; Matsushita, T.; Kikuchi, H.; Isobe, N.; Ohyagi, Y.; Kira, J.-I. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J. Neuroimmunol. 2010, 222, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.M.; Freeman, W.M.; Randazzo, W.T.; Stephens, H.E.; Beard, J.L.; Simmons, Z.; Connor, J.R. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 2008, 72, 14–19. [Google Scholar] [CrossRef]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflammation 2014, 11, 94. [Google Scholar] [CrossRef]
- Delaby, C.; Alcolea, D.; Carmona-Iragui, M.; Illán-Gala, I.; Morenas-Rodríguez, E.; Barroeta, I.; Altuna-Azkargorta, M.; Estellés, T.; Santos-Santos, M.; Turon-Sans, J.; et al. Differential levels of Neurofilament Light protein in cerebrospinal fluid in patients with a wide range of neurodegenerative disorders. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Rossi, D.; Volanti, P.; Brambilla, L.; Colletti, T.; Spataro, R.; La Bella, V. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J. Neurol. 2018, 265, 510–521. [Google Scholar] [CrossRef]
- Olsson, B.; Portelius, E.; Cullen, N.C.; Sandelius, C.; Zetterberg, H.; Andreasson, U.; Höglund, K.; Irwin, D.; Grossman, M.; Weintraub, D.; et al. Association of Cerebrospinal Fluid Neurofilament Light Protein Levels With Cognition in Patients With Dementia, Motor Neuron Disease, and Movement Disorders. JAMA Neurol. 2019, 76, 318–325. [Google Scholar] [CrossRef]
- De Schaepdryver, M.; Jeromin, A.; Gille, B.; Claeys, K.; Herbst, V.; Brix, B.; Van Damme, P.; Poesen, K. Comparison of elevated phosphorylated neurofilament heavy chains in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 89, 367–373. [Google Scholar] [CrossRef]
- Huang, J.; Khademi, M.; Fugger, L.; Lindhe, H.; Novakova, L.; Axelsson, M.; Malmeström, C.; Constantinescu, C.; Lycke, J.; Piehl, F.; et al. Inflammation-related plasma and CSF biomarkers for multiple sclerosis. Proc. Natl. Acad. Sci. 2020, 117, 12952–12960. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Kelley, J.L.; Yerra, L.; Smith, J.K.; Chi, D.S. Human Endothelium as a Source of Multifunctional Cytokines: Molecular Regulation and Possible Role in Human Disease. J. Interf. Cytokine Res. 1999, 19, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.; Pakkenberg, B.; Stark, A. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Hanisch, U.-K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The Good, The Bad, and The Resting. J. Neuroimmune Pharmacol. 2009, 4, 389–398. [Google Scholar] [CrossRef]
- Sawada, M. Neuroprotective and toxic changes in microglia in neurodegenerative disease. Park. Relat. Disord. 2009, 15, S39–S41. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef]
- Graves, M.C.; Fiala, M.; Dinglasan, L.A.V.; Liu, N.Q.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. 2004, 5, 213–219. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2003, 55, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Stow, J.L.; Low, P.C.; Offenhäuser, C.; Sangermani, D. Cytokine secretion in macrophages and other cells: Pathways and mediators. Immunobiology 2009, 214, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Yuen, E.C.; Ransom, B.R.; Möller, T. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 2004, 48, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhao, W.; Beers, D.R.; Yen, A.A.; Xie, W.; Henkel, J.S.; Appel, S.H. Mutant SOD1G93Amicroglia are more neurotoxic relative to wild-type microglia. J. Neurochem. 2007, 102, 2008–2019. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Ishigaki, S.; Yamamoto, M.; Liang, Y.; Niwa, J.-I.; Takeuchi, H.; Doyu, M.; Sobue, G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 80, 158–167. [Google Scholar] [CrossRef]
- Moreau, C.; Devos, D.; Brunaud-Danel, V.; Defebvre, L.; Perez, T.; Destee, A.; Tonnel, A.B.; Lassalle, P.; Just, N. Elevated IL-6 and TNF- levels in patients with ALS: Inflammation or hypoxia? Neurology 2005, 65, 1958–1960. [Google Scholar] [CrossRef]
- Tanaka, M.; Kikuchi, H.; Ishizu, T.; Minohara, M.; Osoegawa, M.; Motomura, M.K.; Tateishi, T.; Ohyagi, Y.; Kira, J.-I. Intrathecal Upregulation of Granulocyte Colony Stimulating Factor and Its Neuroprotective Actions on Motor Neurons in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2006, 65, 816–825. [Google Scholar] [CrossRef][Green Version]
- Almer, G.; Teismann, P.; Stevic, Z.; Halaschek–Wiener, J.; Deecke, L.; Kostic, V.; Przedborski, S. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology 2002, 58, 1277–1279. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.; Sanberg, P.R. Impaired blood–brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of Compromised Blood-Spinal Cord Barrier in Early and Late Symptomatic SOD1 Mice Modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Li, X.; Wang, H.; Wang, T. Elevated cerebrospinal fluid homocysteine is associated with blood-brain barrier disruption in amyotrophic lateral sclerosis patients. Neurol. Sci. 2020, 41, 1865–1872. [Google Scholar] [CrossRef]
- Traynor, B.J.; Codd, M.B.; Corr, B.; Forde, C.; Frost, E.; Hardiman, O. Amyotrophic Lateral Sclerosis Mimic Syndromes. Arch. Neurol. 2000, 57, 109–113. [Google Scholar] [CrossRef]
- Miyashiro, A.; Matsui, N.; Shimatani, Y.; Nodera, H.; Izumi, Y.; Kuwabara, S.; Imai, T.; Baba, M.; Komori, T.; Sonoo, M.; et al. Are multifocal motor neuropathy patients underdiagnosed? An epidemiological survey in Japan. Muscle Nerve 2013, 49, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Breville, G.; Lascano, A.M.; Roux-Lombard, P.; Lalive, P.H. IL-8 as a potential biomarker in Guillain-Barre Syndrome. Eur. Cytokine Netw. 2019, 30, 130–134. [Google Scholar]
- Sainaghi, P.P.; Collimedaglia, L.; Alciato, F.; Leone, M.; Naldi, P.; Molinari, R.; Monaco, F.; Avanzi, G.C. The expression pattern of inflammatory mediators in cerebrospinal fluid differentiates Guillain–Barré syndrome from chronic inflammatory demyelinating polyneuropathy. Cytokine 2010, 51, 138–143. [Google Scholar] [CrossRef]
- Pietikäinen, A.; Maksimow, M.; Kauko, T.; Hurme, S.; Salmi, M.; Hytönen, J. Cerebrospinal fluid cytokines in Lyme neuroborreliosis. J. Neuroinflammation 2016, 13, 1–10. [Google Scholar] [CrossRef]
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of Transactivation-Responsive DNA-Binding Protein 43 and Huntingtin in Inclusions of Huntington Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef]
- Feneberg, E.; Gray, E.; Ansorge, O.; Talbot, K.; Turner, M. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol. Neurobiol. 2018, 55, 7789–7801. [Google Scholar] [CrossRef]
- Gillespie, M.; Stein, R. The relationship between axon diameter, myelin thickness and conduction velocity during atrophy of mammalian peripheral nerves. Brain Res. 1983, 259, 41–56. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Mometto, N.; Bartoletti-Stella, A.; Polischi, B.; Oppi, F.; Poda, R.; Stanzani-Maserati, M.; Cortelli, P.; Liguori, R.; Capellari, S.; et al. Cerebrospinal Fluid Biomarkers in Patients with Frontotemporal Dementia Spectrum: A Single-Center Study. J. Alzheimer’s Dis. 2018, 66, 551–563. [Google Scholar] [CrossRef]
- Skillbäck, T.; Mattsson-Carlgren, N.; Blennow, K.; Zetterberg, H. Cerebrospinal fluid neurofilament light concentration in motor neuron disease and frontotemporal dementia predicts survival. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Skillbäck, T.; Farahmand, B.; Bartlett, J.W.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology 2014, 83, 1945–1953. [Google Scholar] [CrossRef]
- Benatar, M.; Turner, M.R.; Wuu, J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 303–309. [Google Scholar] [CrossRef]
- Boylan, K.B.; Glass, J.D.; E Crook, J.; Yang, C.; Thomas, C.S.; Desaro, P.; Johnston, A.; Overstreet, K.; Kelly, C.; Polak, M.; et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 84, 467–472. [Google Scholar] [CrossRef]
- Feneberg, E.; Oeckl, P.; Steinacker, P.; Verde, F.; Barro, C.; Van Damme, P.; Gray, E.; Grosskreutz, J.; Jardel, C.; Kuhle, J.; et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology 2017, 90, e22–e30. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.-Y.; Lv, G.-P.; Gao, L.-N.; Lu, Y.; Guo, J.; Zang, D.-W. Neurofilament Subunit L Levels in the Cerebrospinal Fluid and Serum of Patients with Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2018, 18, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Macdonald-Wallis, C.; Gray, E.; Pearce, N.; Petzold, A.; Norgren, N.; Giovannoni, G.; Fratta, P.; Sidle, K.; Fish, M.; et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015, 84, 2247–2257. [Google Scholar] [CrossRef]
- Sun, Q.; Zhao, X.; Li, S.; Yang, F.; Wang, H.; Cui, F.; Huang, X. CSF Neurofilament Light Chain Elevation Predicts ALS Severity and Progression. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Hadden, R.D.M.; Cornblath, D.R.; Hughes, R.A.C.; Zielasek, J.; Hartung, H.-P.; Toyka, K.V.; Swan, A.V. The Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group Electrophysiological classification of guillain-barré syndrome: Clinical associations and outcome. Ann. Neurol. 1998, 44, 780–788. [Google Scholar] [CrossRef]
- Hughes, R.; Newsom-Davis, J.; Perkin, G.; Pierce, J. Controlled trial of prednisolone in acute polyneuropathy. Lancet 1978, 312, 750–753. [Google Scholar] [CrossRef]
- Kleyweg, R.P.; Van Der Meché, F.G.A.; Schmitz, P.I.M. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain-Barré syndrome. Muscle Nerve 1991, 14, 1103–1109. [Google Scholar] [CrossRef]
- Merkies, I. Getting closer to patients: The INCAT Overall Disability Sum Score relates better to patients’ own clinical judgement in immune-mediated polyneuropathies. J. Neurol. Neurosurg. Psychiatry 2006, 77, 970–972. [Google Scholar] [CrossRef]
- Petzold, A.; Hinds, N.; Murray, N.F.; Hirsch, N.P.; Grant, D.; Keir, G.; Thompson, E.J.; Reilly, M.M. CSF neurofilament levels: A potential prognostic marker in Guillain-Barre syndrome. Neurology 2006, 67, 1071–1073. [Google Scholar] [CrossRef]
- Petzold, A.; Brettschneider, J.; Jin, K.; Keir, G.; Murray, N.; Hirsch, N.; Itoyama, Y.; Reilly, M.; Takeda, A.; Tumani, H. CSF protein biomarkers for proximal axonal damage improve prognostic accuracy in the acute phase of Guillain-Barré syndrome. Muscle Nerve 2009, 40, 42–49. [Google Scholar] [CrossRef]
- Wang, X.-K.; Zhang, H.; Meng, F.-H.; Chang, M.; Wang, Y.-Z.; Jin, T.; Mix, E.; Zhu, J. Elevated levels of S100B, tau and pNFH in cerebrospinal fluid are correlated with subtypes of Guillain–Barré syndrome. Neurol. Sci. 2012, 34, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Dujmovic, I.; Lunn, M.P.; Reilly, M.M.; Petzold, A. Serial cerebrospinal fluid neurofilament heavy chain levels in severe Guillain-Barré syndrome. Muscle Nerve 2012, 48, 132–134. [Google Scholar] [CrossRef]
- Axelsson, M.; Sjögren, M.; Andersen, O.; Blennow, K.; Zetterberg, H.; Lycke, J. Neurofilament light protein levels in cerebrospinal fluid predict long-term disability of Guillain-Barré syndrome: A pilot study. Acta Neurol. Scand. 2018, 138, 143–150. [Google Scholar] [CrossRef]
- Mariotto, S.; Farinazzo, A.; Magliozzi, R.; Alberti, D.; Monaco, S.; Ferrari, S. Serum and cerebrospinal neurofilament light chain levels in patients with acquired peripheral neuropathies. J. Peripher. Nerv. Syst. 2018, 23, 174–177. [Google Scholar] [CrossRef]
- Körtvelyessy, P.; Kuhle, J.; Düzel, E.; Vielhaber, S.; Schmidt, C.; Heinius, A.; Leypoldt, F.; Schraven, B.; Reinhold, D.; Leppert, D.; et al. Ratio and index of Neurofilament light chain indicate its origin in Guillain-Barré Syndrome. Ann. Clin. Transl. Neurol. 2020, 7, 2213–2220. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G.; Pratt, C.W. Principles of Biochemistry, 3rd. ed.; International Student Version; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Capodivento, G.; Visigalli, D.; Garnero, M.; Fancellu, R.; Ferrara, M.D.; Basit, A.; Hamid, Z.; Pastore, V.P.; Garibaldi, S.; Armirotti, A.; et al. Sphingomyelin as a myelin biomarker in CSF of acquired demyelinating neuropathies. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Capodivento, G.; De Michelis, C.; Carpo, M.; Fancellu, R.; Schirinzi, E.; Severi, D.; Visigalli, D.; Franciotta, D.; Manganelli, F.; Siciliano, G.; et al. CSF sphingomyelin: A new biomarker of demyelination in the diagnosis and management of CIDP and GBS. J. Neurol. Neurosurg. Psychiatry 2020, 92, 303–310. [Google Scholar] [CrossRef]
- Péter, M.; Török, W.; Petrovics-Balog, A.; Vígh, L.; Vécsei, L.; Balogh, G. Cerebrospinal fluid lipidomic biomarker signatures of demyelination for multiple sclerosis and Guillain–Barré syndrome. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Newman, D.J. Cystatin C. Ann. Clin. Biochem. Int. J. Lab. Med. 2002, 39, 89–104. [Google Scholar] [CrossRef]
- Knight, E.L.; Verhave, J.C.; Spiegelman, D.; Hillege, H.L.; De Zeeuw, D.; Curhan, G.C.; De Jong, P.E. Factors influencing serum cystatin C levels other than renal function and the impact on renal function measurement. Kidney Int. 2004, 65, 1416–1421. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, S.; Qin, Z.; Cui, Y.; Qin, Y.; Bai, S. Alteration of cystatin C levels in cerebrospinal fluid of patients with Guillain-Barré Syndrome by a proteomical approach. Mol. Biol. Rep. 2008, 36, 677–682. [Google Scholar] [CrossRef]
- Yang, Y.-R.; Liu, S.-L.; Qin, Z.-Y.; Liu, F.-J.; Qin, Y.-J.; Bai, S.-M.; Chen, Z.-Y. Comparative Proteomics Analysis of Cerebrospinal Fluid of Patients with Guillain–Barré Syndrome. Cell. Mol. Neurobiol. 2008, 28, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, S.; Zhang, R.; Pei, J.; Chen, L.; Cao, Y.; Zhang, H.; Yang, G. Identification of CSF biomarkers by proteomicsin Guillain-Barr�syndrome. Exp. Ther. Med. 2018, 15, 5177–5182. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Uemichi, T. Transthyretin amyloidosis. Amyloid 1996, 3, 44–56. [Google Scholar] [CrossRef]
- Jin, T.; Hu, L.-S.; Chang, M.; Wu, J.; Winblad, B.; Zhu, J. Proteomic identification of potential protein markers in cerebrospinal fluid of GBS patients. Eur. J. Neurol. 2007, 14, 563–568. [Google Scholar] [CrossRef]
- Chiang, H.-L.; Lyu, R.-K.; Tseng, M.-Y.; Chang, K.-H.; Chang, H.-S.; Hsu, W.-C.; Kuo, H.-C.; Chu, C.-C.; Wu, Y.-R.; Ro, L.-S.; et al. Analyses of transthyretin concentration in the cerebrospinal fluid of patients with Guillain-Barré syndrome and other neurological disorders. Clin. Chim. Acta 2009, 405, 143–147. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Zhang, X.-M.; Mao, X.-J.; Deng, H.; Li, H.-F.; Press, R.; Fredrikson, S.; Zhu, J. Altered cerebrospinal fluid index of prealbumin, fibrinogen, and haptoglobin in patients with Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. Acta Neurol. Scand. 2011, 125, 129–135. [Google Scholar] [CrossRef]
- Chang, K.-H.; Lyu, R.-K.; Tseng, M.-Y.; Ro, L.-S.; Wu, Y.-R.; Chang, H.-S.; Hsu, W.-C.; Kuo, H.-C.; Huang, C.-C.; Chu, C.-C.; et al. Elevated haptoglobin level of cerebrospinal fluid in Guillain-Barré syndrome revealed by proteomics analysis. Proteom. Clin. Appl. 2007, 1, 467–475. [Google Scholar] [CrossRef]
- Lehmensiek, V.; Süssmuth, S.; Brettschneider, J.; Tauscher, G.; Felk, S.; Gillardon, F.; Tumani, H. Proteome analysis of cerebrospinal fluid in Guillain–Barré syndrome (GBS). J. Neuroimmunol. 2007, 185, 190–194. [Google Scholar] [CrossRef]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Reiber, H.; Tumani, H. Tau protein in cerebrospinal fluid (CSF): A blood–CSF barrier related evaluation in patients with various neurological diseases. Neurosci. Lett. 2001, 300, 95–98. [Google Scholar] [CrossRef]
- Jin, K.; Takeda, A.; Shiga, Y.; Sato, S.; Ohnuma, A.; Nomura, H.; Arai, H.; Kusunoki, S.; Ikeda, M.; Itoyama, Y. CSF tau protein: A new prognostic marker for Guillain-Barre syndrome. Neurology 2006, 67, 1470–1472. [Google Scholar] [CrossRef]
- Blanc, F.; Jaulhac, B.; Fleury, M.; de Seze, J.; de Martino, S.J.; Remy, V.; Blaison, G.; Hansmann, Y.; Christmann, D.; Tranchant, C. Relevance of the antibody index to diagnose Lyme neuroborreliosis among seropositive patients. Neurology 2007, 69, 953–958. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Vitte, J.; Fassier, C.; Tiziano, F.D.; Dalard, C.; Soave, S.; Roblot, N.; Brahe, C.; Saugier-Veber, P.; Bonnefont, J.P.; Melki, J. Refined Characterization of the Expression and Stability of the SMN Gene Products. Am. J. Pathol. 2007, 171, 1269–1280. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2014, 51, 157–167. [Google Scholar] [CrossRef]
- Singh, R.N.; Ottesen, E.W.; Singh, N.N. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef]
- Hegen, H.; Auer, M.; Zeileis, A.; Deisenhammer, F. Upper reference limits for cerebrospinal fluid total protein and albumin quotient based on a large cohort of control patients: Implications for increased clinical specificity. Clin. Chem. Lab. Med. 2016, 54, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J. Neurochem. 2020, 153, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Müschen, L.; Osmanovic, A.; Binz, C.; Jendretzky, K.; Ranxha, G.; Bronzlik, P.; Abu-Fares, O.; Wiehler, F.; Möhn, N.; Hümmert, M.; et al. Cerebrospinal Fluid Parameters in Antisense Oligonucleotide-Treated Adult 5q-Spinal Muscular Atrophy Patients. Brain Sci. 2021, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Koch, J.C.; Cordts, I.; Dreyhaupt, J.; Otto, M.; Uzelac, Z.; Witzel, S.; Winter, B.; Kocak, T.; Schocke, M.; et al. Routine Cerebrospinal Fluid (CSF) Parameters in Patients With Spinal Muscular Atrophy (SMA) Treated With Nusinersen. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- McCudden, C.R.; Brooks, J.; Figurado, P.; Bourque, P.R. Cerebrospinal Fluid Total Protein Reference Intervals Derived from 20 Years of Patient Data. Clin. Chem. 2017, 63, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Gingele, S.; Hümmert, M.W.; Alvermann, S.; Jendretzky, K.F.; Bönig, L.; Brieskorn, M.; Schwenkenbecher, P.; Sühs, K.-W.; Müschen, L.H.; Osmanovic, A.; et al. Routine Cerebrospinal Fluid Cytology Reveals Unique Inclusions in Macrophages During Treatment With Nusinersen. Front. Neurol. 2019, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- Machacek, M.E.; Gogakos, T.; Fletcher, M.C.; Lunderville, K.A.; Swoboda, K.J.; Sohani, A.R. Unusual inclusions in cerebrospinal fluid macrophages of spinal muscular atrophy patients treated with nusinersen. Int. J. Lab. Hematol. 2020. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, B.H.; Yip, W.; Chou, P.; Yip, B.-S. Neurofilament Proteins as Prognostic Biomarkers in Neurological Disorders. Curr. Pharm. Des. 2020, 25, 4560–4569. [Google Scholar] [CrossRef]
- Abdelhak, A.; Huss, A.; Kassubek, J.; Tumani, H.; Otto, M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nat. Cell Biol. 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Shoji, M. Cerebrospinal Fluid and Plasma Tau as a Biomarker for Brain Tauopathy. Tau Biol. 2019, 1184, 393–405. [Google Scholar] [CrossRef]
- Introna, A.; Milella, G.; D’Errico, E.; Fraddosio, A.; Scaglione, G.; Ucci, M.; Bsc, M.R.; Simone, I.L. Is cerebrospinal fluid amyloid-β42 a promising biomarker of response to nusinersen in adult spinal muscular atrophy patients? Muscle Nerve 2021, 63, 905–909. [Google Scholar] [CrossRef]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.-S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.-K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef]
- Winter, B.; Guenther, R.; Ludolph, A.C.; Hermann, A.; Otto, M.; Wurster, C.D. Neurofilaments and tau in CSF in an infant with SMA type 1 treated with nusinersen. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1068–1069. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein Are Not Elevated in Cerebrospinal Fluid of Adult Patients with Spinal Muscular Atrophy during Loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef]
- Vågberg, M.; Norgren, N.; Dring, A.; Lindqvist, T.; Birgander, R.; Zetterberg, H.; Svenningsson, A. Levels and Age Dependency of Neurofilament Light and Glial Fibrillary Acidic Protein in Healthy Individuals and Their Relation to the Brain Parenchymal Fraction. PLoS ONE 2015, 10, e0135886. [Google Scholar] [CrossRef] [PubMed]
- Gafson, A.R.; Barthélemy, N.R.; Bomont, P.; O Carare, R.; Durham, H.D.; Julien, J.-P.; Kuhle, J.; Leppert, D.; A Nixon, R.; O Weller, R.; et al. Neurofilaments: Neurobiological foundations for biomarker applications. Brain 2020, 143, 1975–1998. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef]
- Wurster, C.D.; Günther, R.; Steinacker, P.; Dreyhaupt, J.; Wollinsky, K.; Uzelac, Z.; Witzel, S.; Kocak, T.; Winter, B.; Koch, J.C.; et al. Neurochemical markers in CSF of adolescent and adult SMA patients undergoing nusinersen treatment. Ther. Adv. Neurol. Disord. 2019, 12. [Google Scholar] [CrossRef] [PubMed]


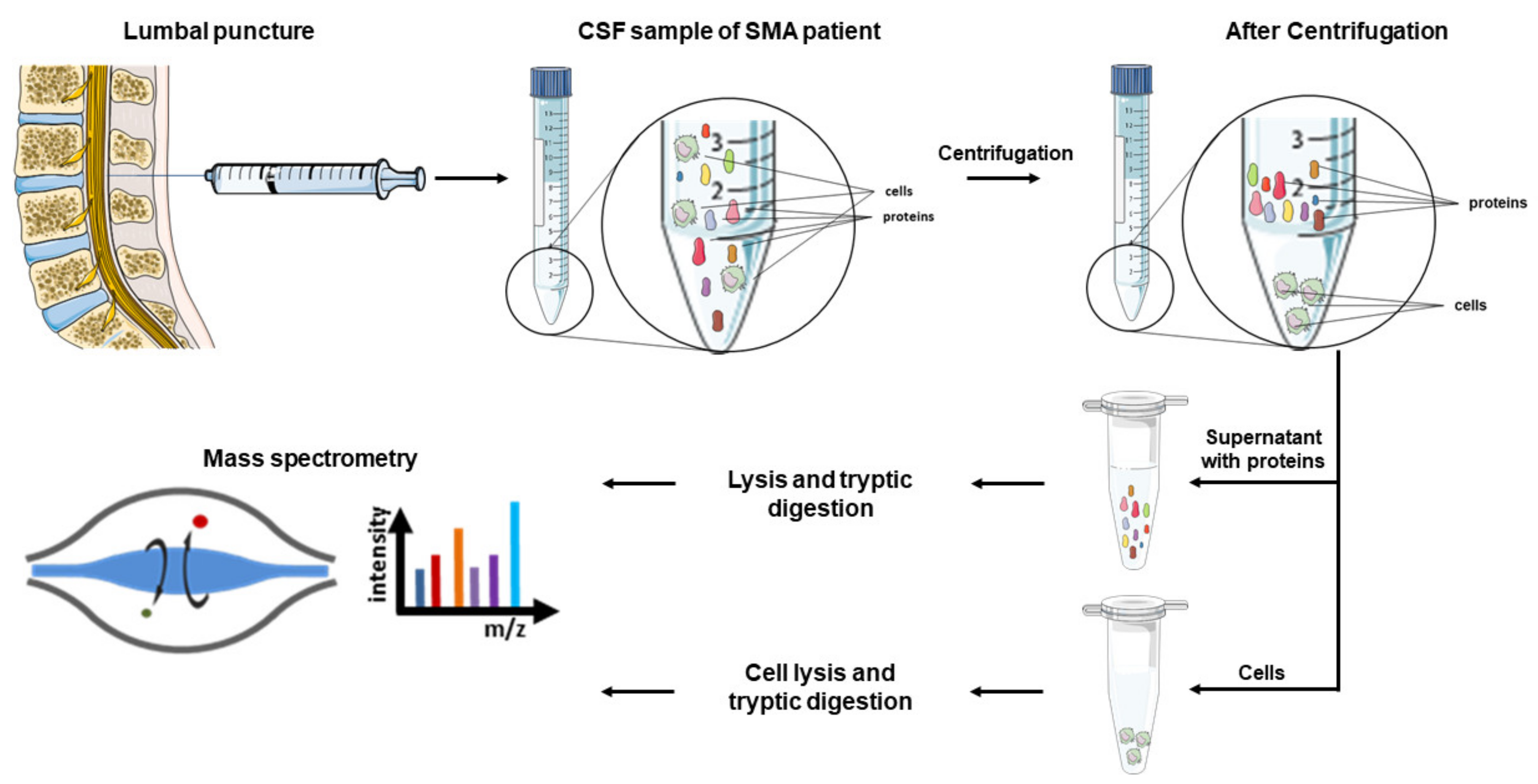{kind=link}
| Cytokines | Tateishi et al. | Mitchell et al. | Furukawa et al. | Italiani et al. | Breville et al. | Sainaghi et al. | Pietikäinen et al. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALS/ONDs | ALS/ONDs | ALS/ONDs | ALS/MMN | PMA/ONDs | PMA/MMN | MMN/ONDs | ALS/ ONDs | GBS/CIDP | GBS/NIP | GBS/CIDP | GBS/Control | CIDP/Control | LNB/Control | LNB/MS | LNB/TBE | |
| IL-1α | - | - | - | - | - | - | - | - | - | - | - | - | - | x | √ | x |
| IL-1β | √ | x | x | x | x | x | x | x | - | - | n. e. | n. e. | n. e. | √ | √ | x |
| IL-1ra | x | x | x | x | √ | x | √ | x | - | - | √ | √ | x | √ | √ | x |
| IL-2 | x | √ | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | n. e. | n. e. | n. e. |
| IL-2ra | - | - | - | - | - | - | - | - | - | - | x | x | x | x | x | x |
| IL-3 | - | - | - | - | - | - | - | - | - | - | - | - | - | √ | √ | x |
| IL-4 | x | x | √ | √ | x | x | x | - | - | - | n. e. | n. e. | n. e. | √ | x | √ |
| IL-5 | x | x | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | n. e. | n. e. | n. e. |
| IL-6 | x | √ | x | x | x | x | x | - | x | x | x | x | x | √ | √ | √ |
| IL-7 | √ | x | √ | x | √ | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | √ |
| IL-9 | √ | x | x | x | x | x | x | - | - | - | x | x | x | √ | √ | x |
| IL-10 | x | x | x | x | √ | √ | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | √ |
| IL-12 | √ | x | x | x | x | x | x | - | - | - | x | x | x | √ | √ | √ |
| IL-13 | x | x | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | x |
| IL-15 | x | √ | x | x | x | x | x | - | - | - | x | x | x | x | x | √ |
| IL-16 | - | - | - | - | - | - | - | - | - | - | x | x | x | √ | √ | x |
| IL-17 | √ | √ | √ | √ | √ | x | x | - | - | - | n. e. | n. e. | n. e. | √ | x | √ |
| TNF-α | √ | x | x | x | x | x | x | - | x | x | n. e. | n. e. | n. e. | √ | √ | √ |
| TNF-β | - | - | - | - | - | - | - | - | - | - | - | - | - | x | x | x |
| IFNα-2 | - | - | - | - | - | - | - | - | - | - | x | x | x | √ | x | x |
| IFN-y | √ | x | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | √ |
| CCL2 | √ | √ | x | x | x | x | x | - | - | - | x | √ | √ | x | x | x |
| CCL3 | x | √ | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | x |
| CCL4 | √ | √ | x | x | x | x | x | - | - | - | x | x | x | - | - | - |
| CCL5 | √ | x | x | x | x | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | x |
| CCL7 | - | - | - | - | - | - | - | - | - | - | √ | √ | √ | n. e. | n. e. | n. e. |
| CCL11 | √ | x | √ | x | √ | x | x | - | - | - | n. e. | n. e. | n. e. | √ | √ | √ |
| CCL27 | - | - | - | - | - | - | - | - | - | - | x | √ | √ | √ | √ | x |
| CXCL1 | - | - | - | - | - | - | - | - | - | - | x | x | x | √ | √ | x |
| CXCL8 | √ | x | x | x | x | x | x | - | √ | √ | √ | √ | x | √ | √ | x |
| CXCL9 | - | - | - | - | - | - | - | - | - | - | x | √ | √ | √ | √ | x |
| CXCL10 | √ | x | x | x | x | x | x | - | - | - | √ | √ | √ | √ | √ | x |
| CXCL 12 | - | - | - | - | - | - | - | - | - | - | x | √ | √ | - | - | - |
| CXCL 12α | - | - | - | - | - | - | - | - | - | - | - | - | - | √ | √ | x |
| CXCL13 | - | - | - | - | - | - | - | - | - | - | - | - | - | √ | √ | √ |
| G-CSF | √ | √ | √ | √ | √ | √ | x | - | - | - | x | x | x | √ | √ | √ |
| GM-CSF | x | √ | x | x | x | x | x | - | - | - | x | x | x | √ | √ | x |
| M-CSF | - | - | - | - | - | - | - | - | - | - | x | x | x | x | x | x |
| FGF-2 | x | √ | √ | √ | √ | √ | x | - | - | - | x | x | x | √ | √ | √ |
| PDGFbb | x | x | √ | x | √ | x | x | - | - | - | x | x | x | √ | √ | x |
| VEGF | √ | √ | x | x | √ | √ | x | - | - | - | x | √ | √ | √ | √ | x |
| β2M | - | x | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| TNFR1 | - | - | x | x | √ | x | x | - | - | - | - | - | - | - | - | - |
| sIL-1R2 | - | - | - | - | - | - | - | x | - | - | - | - | - | - | - | - |
| IL-33 | - | - | - | - | - | - | - | x | - | - | - | - | - | - | - | - |
| sIL-1R4 | - | - | - | - | - | - | - | x | - | - | - | - | - | - | - | - |
| IL-37 | - | - | - | - | - | - | - | x | - | - | - | - | - | - | - | - |
| IL-18 | - | - | - | - | - | - | - | √ | - | - | x | x | x | √ | √ | x |
| IL-18BP | - | - | - | - | - | - | - | √ | - | - | - | - | - | - | - | - |
| ICAM-1 | - | - | - | - | - | - | - | - | - | - | x | √ | √ | - | - | - |
| VCAM1 | - | - | - | - | - | - | - | - | - | - | x | √ | √ | - | - | - |
| SCF | - | - | - | - | - | - | - | - | - | - | √ | x | √ | √ | √ | x |
| LIF | - | - | - | - | - | - | - | - | - | - | x | x | x | x | x | x |
| MIF | - | - | - | - | - | - | - | - | - | - | x | x | x | x | √ | x |
| SCGF-b | - | - | - | - | - | - | - | - | - | - | x | x | x | x | √ | x |
| HGF | - | - | - | - | - | - | - | - | - | - | √ | x | √ | x | x | x |
| TRAIL | - | - | - | - | - | - | - | - | - | - | x | x | x | √ | √ | x |
| β-NGF | - | - | - | - | - | - | - | - | - | - | - | - | - | √ | √ | x |
| Paper | Cohort Size and Composition | Body Fluid | Marker | Method for TDP-43 | Antibody | Results TDP-43 ng/mL |
|---|---|---|---|---|---|---|
| Kasai et al., 2009 | 30 ALS, 29 controls (13 controls, 16 disease controls) | CSF | TDP-43 | Sandwich ELISA (Nunc MaxiSorp, Xat-bottom 96-well Black MicroWell plate, Roskilde, Denmark) | anti-TDP-43 monoclonal antibody, detection antibody, anti-TDP-43 rabbit polyclonal antibody (10782-2-AP, ProteinTech Group, Chicago, IL, USA), raised against a recombinant protein corresponding to residues 1–261 of human TDP-43 (H00023435-M01, clone 2E2-D3, Abnova Corporation, Walnut, CA, USA), detection antibody, anti-TDP-43 rabbit polyclonal antibody (10782-2-AP, ProteinTech Group, Chicago, IL, USA) | ALS: 6.92 +/− 3.71; Control: 5.31 +/− 0.94 |
| Noto et al., 2010 | 27 ALS, 50 disease controls (15 PD, 15 MS, 20 GBS) | CSF | TDP-43 | Sandwich ELISA (Nunc MaxiSorp, Xat-bottom 96-well Black MicroWell plate, Roskilde, Denmark) | anti-TDP-43 monoclonal antibody (H00023435-M01, clone 2E2-D3, Abnova Corporation, Walnut, CA, USA), detection antibody, anti-TDP-43 rabbit polyclonal antibody (10782-2-AP, ProteinTech Group, Chicago, IL, USA) | ALS: 29.5 +/− 15.5; Control PD: 19.7 +/− 6.6, Control MS: 13.7 +/− 9.0, Control GBS: 16.7 +/− 7.5 |
| Hosogawa et al., 2014 | 13 ALS, 7 GBS | CSF | TDP-43 | Sandwich ELISA (Nunc MaxiSorp, Xat-bottom 96-well Black MicroWell plate, Roskilde, Denmark) | monoclonal antibody, clone 2E2-D3 (Abnova Corp., Taipei, Taiwan), for capture and a rabbit polyclonal antibody (catalog code 10782-2-AP, ProteinTech Group Inc., Chicago, IL, USA) for detection. | ALS: 1.68 +/− 0.15; GBS: 1.05 +/− 0.13 |
| Steinacker et al., 2008 | 15 ALS, 12 FTLD, 9 ALS + FTLD, 3 ALS + frontal disinhibition, 13 controls | CSF | TDP-43 | Immunoblot | Affinity purified polyclonal rabbit antibody raised against amino acids 1 through 260 of recombinant TDP-43 (1:2000 and 1:10,000 to 1:1000; Proteintech Group Inc, Chicago, IL, USA), Monoclonal TDP-43 antibody clone 2E2-D3 specific for amino acids 205 through 25517 (1:1000; Abnova, Taipei City, Taiwan). Polyclonal rabbit antisera raised against N-terminus amino acids 6 through 24 or against C-terminus amino acids 396 through 414 of TDP-43 (1:5000 for both) | not applicable |
| Feneberg et al., 2014 | 9 ALS, 4 FTLD, 8 controls | CSF, Plasma and Brain | TDP-43 | Immunoblot | Antibodies against different TDP-43 epitopes (N-terminus, C-terminus, and aa 205–222), against calnexin, GP Ib-V-IX and flotillin-1. Standard (human Jurkat cells and murine neuroblastoma (N2A) cells were used) | not applicable |
| Paper | Cohort Size and Composition | Body Fluid | Marker | Method for NfL, pNfH | Results pNfH pg/mL | Results NfL [pg/mL] |
|---|---|---|---|---|---|---|
| Poesen et al., 2017 | 220 ALS, 316 DC, 50 DM | CSF | NfL, pNfH | ELISA kits for pNfH (Biovendor, Brno, Czech Republic; RD191138300R, average test-retest variance of 6.8%) and NfL (UmanDiagnostics AB, Umea, Sweden; UD51001, average test-retest variance of 4.9%). | ALS: 2366 (114–18,089), DC: 289 (24–18,740) DM: 296 (24–7049) | ALS: 9427 (370–108,909). DC: 1790 (262–53,677), DM: 1407 (613–36,597) |
| Rossi et al., 2018 | 190 ALS, 130 mixed neurological diseases CTRL-1 (non-inflammatory neurological diseases), CTRL-2 (patients with acute/subacute inflammatory diseases and tumors) | CSF | NfL, pNfH | Single-batch ELISA kits for NF-L assays (iMyBioSource San Diego, USA and UmanDiagnostics AB, Umeå, Sweden). For pNF-ELISA kit from BioVendor Research and Diagnostic Product, Czech Republic) | ALS:1700 (760–3170), CTRL 1: 30 (0.00–320) CTRL 2: 820 (0.00–3470) | MyoBioSource kit: ALS: 2140 (1350–3300), CTRL 1: 2040 (1250–3390) CTRL 2: 3090 (1120–4590); UmanDiagnostics kit: ALS: 4700 (760–3170) CTRL 1: 300 (0.00–320) CTRL 2: 820 (0.00–3470) |
| Olsson et al., 2018 | 68 ALS, 75 controls, 114 patients with MCI, 397 AD, 96 FTLD, 41 PD, 19 PD with MCI, 29 with PD dementia, 33 LBD, 21 with CS, 20 with progressive supranuclear palsy (PSP) | CSF | NfL | In-house ELISA (2 NFL mouse monoclonal antibodies (NFL21 and NFL23)) | not tested | ALS: 4185 (2207–7453), CTRL: 536 (398–777), MCI: 831 (526–1075), AD: 951 (758–1261), FTD: 1873 (830–2588), PD: 619 (526–840), PD MCI: 779 (464–1021), PD dementia: 981 (679–1722), DLB: 991 (695–2139), CBS: 1281 (828–2713), PSP: 1578 (1287–3104) |
| Delaby et al., 2020 | 46, ALS, 118 Controls, 116 AD, 47 DS, 56, FTD, 37 DLB, 26 PDLB, 26 CS, 12 PSP | CSF | NfL | ELISA kit (NF-light, UMAN DIAGNOSTICS, Umea, Sweden) | not tested | ALS: 2953 (1664–4250), CTRL: 411 (343–567), AD: 940 (765–1229), DS: 349 (196–464), DS + AD: 955 (664–1497), FTLD: 1240 (859–2378), DLB: 1135 (803–1321), PDLB: 934 (643–1094), CBS: 1637 (923–2797) PSP: 1422 (1034–1727) |
| Oeckl et al., 2016 | Multicenter study (15 centers, 5 ALS patients each (150 ALS patients in total), DC (details on composition in each center can be found in the respective paper) | CSF | NfL, pNfH | ELISA (pNfH: BioVendor GmbH; NfL: IBL International), Two aliqouts per sample One analysed at Neurochemical Laboratory at the Department of Neurology in Ulm and the other at the department of metabolic biochemistry, Hôpitaux Universitaires Pitié Salpêtrière-Charles Foix, Paris. | ALS: 1773.2 (average centers, Ulm), CTRL 476.3 (average centers, Ulm), ALS: 1755.1 (average all centers, Paris), CTRL 288.8 (average centers, Paris) | ALS: 4148.6 (average all centers, Ulm), CTRL 284.2 (average from all centers, Ulm) ALS: 11,577.2 (average all centers, Paris), CTRL 1242.4 (average from all centers, Paris) |
| De Schaepdryver et al., 2017 | 85 ALS, 215 DC, 31 DM | CSF and Plasma | pNfH | ELISA for pNfH concentrations (Euroimmun AG, Lübeck, Germany). ELISA from Biovendor (RD191138300R, Brno, Czech Republic), from 83 patients with ALS and 213 controls were used from the previous publication, 9 to perform a paired method comparison with the IVD ELISA from Euroimmun. | ALS: 2451 (314–17,247); DC: 281 (20–13,669) | not measured |
| Paper | Cohort Size and Composition | Other Criteria | Marker | Method | Results |
|---|---|---|---|---|---|
| Petzold et al., 2006 | 23 GBS patients | Pattern, Outcome | NfH | ELISA | NfH level correlates with axonal degeneration and worse outcome |
| Petzold et al., 2009 | 38 GBS patients, 38 controls with other neurological conditions | Outcome | NfH | in-house developed ELISA. | Higher levels in group with poor outcome. NfH > 0.73 ng/mL predicts poor outcome |
| Wang et al., 2012 | 11 AIDP, 11 AMAN, 10 OND | Pattern, Outcome | pNfH | pNFH ELISA kits from Biovendor, Ostrava, Czech Republic; | pNfH higher in AMAN than in AIDP, Correlation between pNFH and outcome in AMAN |
| Dujmovic et al., 2016 | 3 GBS patients | NfH | ELISA | Higher levels of NfH seem to be associated with a poor outcome | |
| Axelson et al., 2018 | 18 GBS patients | Outcome | NfL | ELISA | Poor outcome in patients with NfL levels over 10,000 ng/L |
| Mariotto et al., 2018 | GBS (N = 5), MMN (N = 3), CIDP and variants (N = 12), anti-MAGneuropathy (N = 3), and non-systemic vasculitic neuropathy (N = 1) | Outcome | NfL | HD-1 immunoassay analyzer, Quanterix SimoaTM | No corellation between CSF NfL and outcome |
| Körtvelyessy et al., 2020 | 21 GBS patients, 19 controls | Pattern, Outcome, CSF/serum-ratio | NfL | ELISA | NfL significantly higher than in controls, GBSDS correlated with CSF-NfL, Ratio distinguishes between patterns |
| Paper | Cohort Size and Composition | Protein Marker | Methods | Results |
|---|---|---|---|---|
| Vagberg et al., 2015 | 53 healthy volunteers | NfL, GFAP | ELISA | NfL and GFAP levels in CSF correlated with age (NfL: rho = 0.870; GFAP: rho = 0.595; p < 0.001) |
| Olsson et al., 2019 | 12 patients with SMA type 1, 11 patients sampled for facial nerve palsy or to exclude meningitis or cerebellitis used as control | NfL, tau, GFAP | ELISA | mean baseline levels for all three markers were significantly increased in SMA 1 patients (NfL: 4598 ± 981 pg/mL vs. 148 ± 39 pg/mL, p = 0.001; tau: 939 ± 159 pg/mL vs. 404 ± 86 pg/mL, p = 0.02; GFAP: 236 ± 44 pg/mL vs. 108 ± 26 pg/mL, p = 0.02), NfL levels normalized <380 pg/mL at T4 or T5, tau and GFAP levels decreased over time, correlation with improvement in CHOP-INTEND score was observed for tau (rho = −0.85, p = 0.0008) and NfL (rho = −0.64, p = 0.03) |
| Totzeck et al., 2019 | 11 patients with SMA type 3 | NfH, tau | ELISA | mean levels for tau and NfH remained stable within the reference range (tau: <290 pg/mL; NfH: <0.69 ng/mL) |
| Winter et al., 2019 | 1 patient with SMA type 1 | NfL, pNfH, total tau, phosphorylated tau | not mentioned | NfL and pNfH level decreased under limit of detection at T4 resepctively T6, total and phosphorylated tau level decreased slightly |
| Wurster et al., 2019 | 9 patients with SMA type 2, 16 with SMA type 3, 25 control patients | NfL, pNfH | ELISA | Median NfL and pNfH levels did not differ significantly from controls at baseline or at T4 |
| Faravelli et al., 2020 | 12 patients with SMA type 3, 9 control patients | NfL, pNfH | ELISA | NfL and pNfH levels were comparable between SMA patients and controls at baseline, NfL and pNfH levels decreased significantly after 6 months of treatment (p = 0.031 respectively p = 0.016) |
| Introna & Milella et al., 2021 | 8 patients with SMA type 2/3 | aβ 40, aβ 42 | solid-phase enzyme immunoassay | mean aβ 40 level remained stable during 420 days of Nusinersen-treatment (T0: 6437.5 ± 3201 pg/mL; T4: 6842.9 ± 1391.5 pg/mL, p = 0.498); mean aβ 42 level increased during treatment, significant at T2 and T4 (T0: 577.3 ± 227 pg/mL; T2: 634.6 ± 266 pg/mL, p = 0.012; T4: 891 ± 462.2 pg/mL, p = 0.018) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krause, K.; Wulf, M.; Sommer, P.; Barkovits, K.; Vorgerd, M.; Marcus, K.; Eggers, B. CSF Diagnostics: A Potentially Valuable Tool in Neurodegenerative and Inflammatory Disorders Involving Motor Neurons: A Review. Diagnostics 2021, 11, 1522. https://doi.org/10.3390/diagnostics11091522
Krause K, Wulf M, Sommer P, Barkovits K, Vorgerd M, Marcus K, Eggers B. CSF Diagnostics: A Potentially Valuable Tool in Neurodegenerative and Inflammatory Disorders Involving Motor Neurons: A Review. Diagnostics. 2021; 11(9):1522. https://doi.org/10.3390/diagnostics11091522
Chicago/Turabian StyleKrause, Karsten, Maximilian Wulf, Paula Sommer, Katalin Barkovits, Matthias Vorgerd, Katrin Marcus, and Britta Eggers. 2021. "CSF Diagnostics: A Potentially Valuable Tool in Neurodegenerative and Inflammatory Disorders Involving Motor Neurons: A Review" Diagnostics 11, no. 9: 1522. https://doi.org/10.3390/diagnostics11091522
APA StyleKrause, K., Wulf, M., Sommer, P., Barkovits, K., Vorgerd, M., Marcus, K., & Eggers, B. (2021). CSF Diagnostics: A Potentially Valuable Tool in Neurodegenerative and Inflammatory Disorders Involving Motor Neurons: A Review. Diagnostics, 11(9), 1522. https://doi.org/10.3390/diagnostics11091522