
Sudden Unexpected Death Associated with Arrhythmogenic Cardiomyopathy: Study of the Cardiac Conduction System



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Data Collection
2.2. Necropsy Investigational Protocol
2.3. Statistical Analysis
3. Results
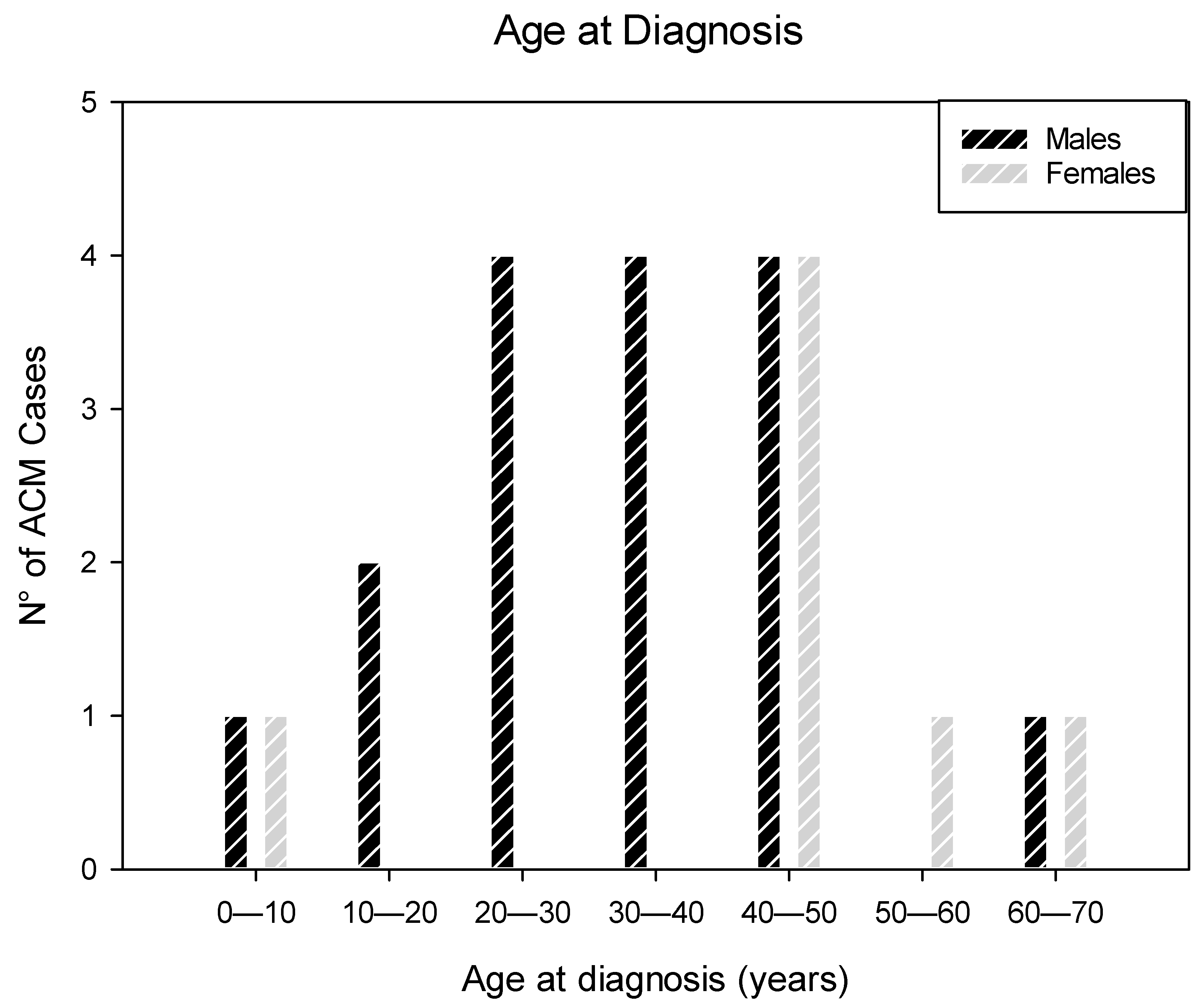
3.1. Patients’ Characteristics
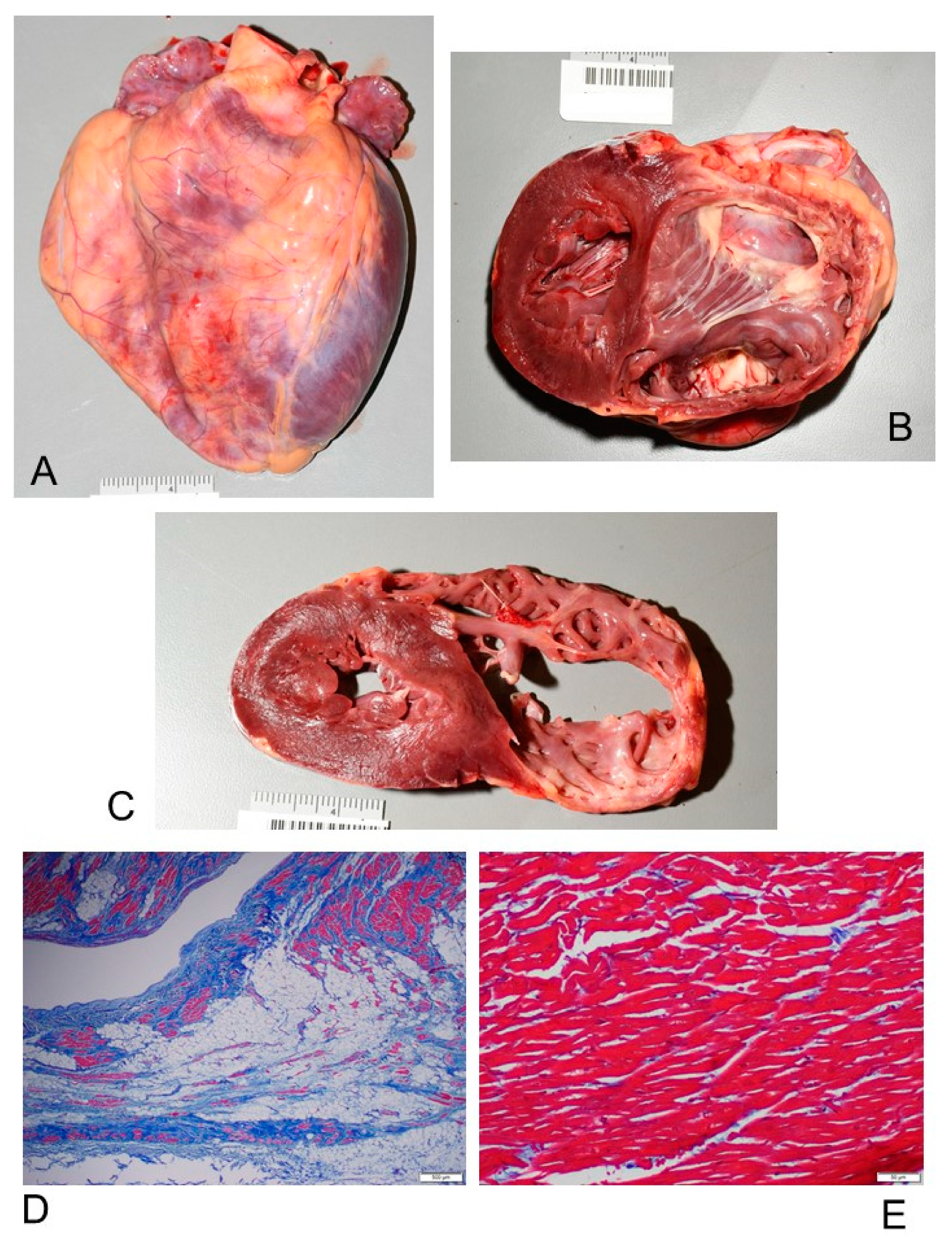
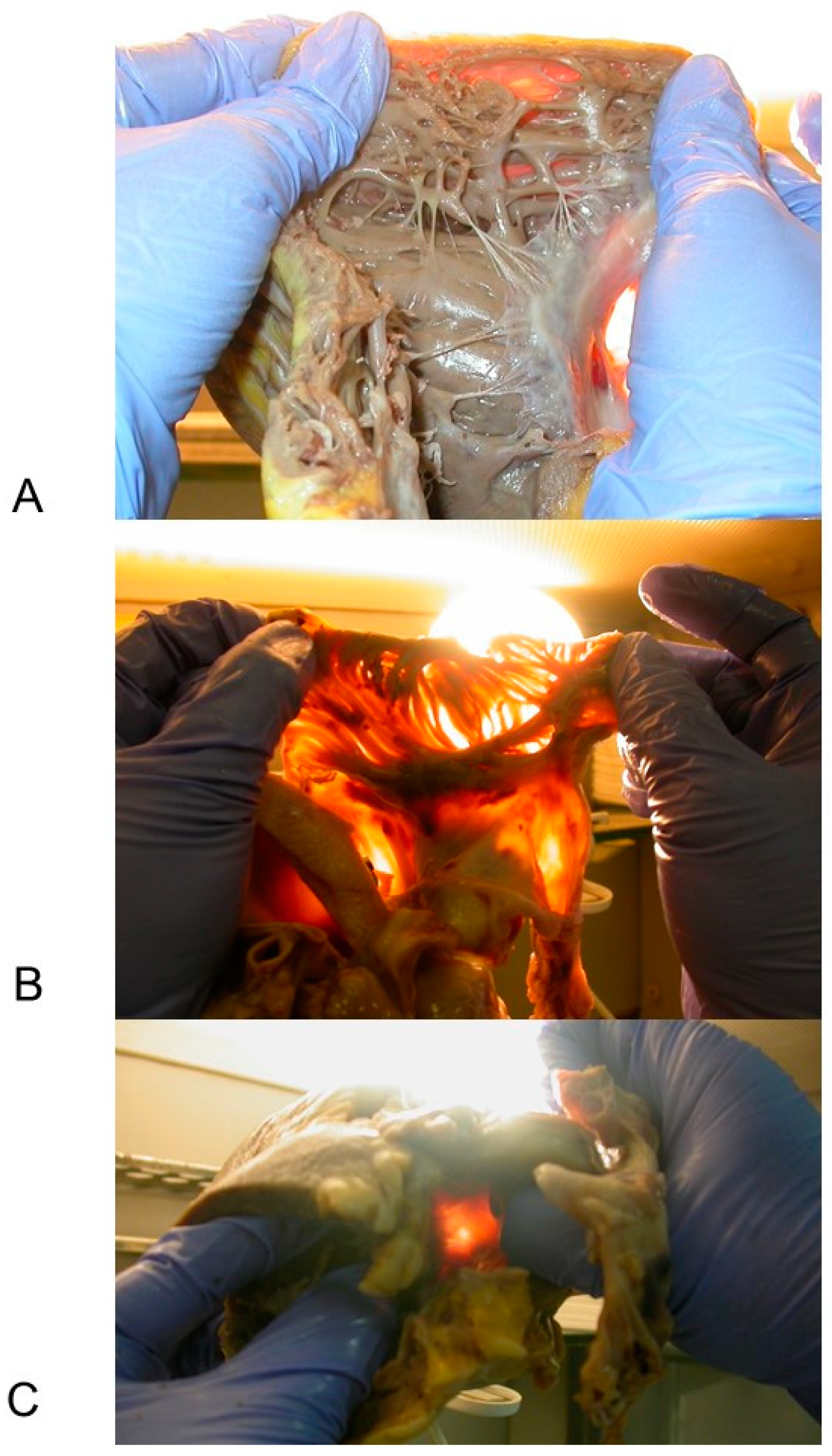
3.2. Anatomo-Pathological Findings of the Heart
3.2.1. Macroscopic Findings
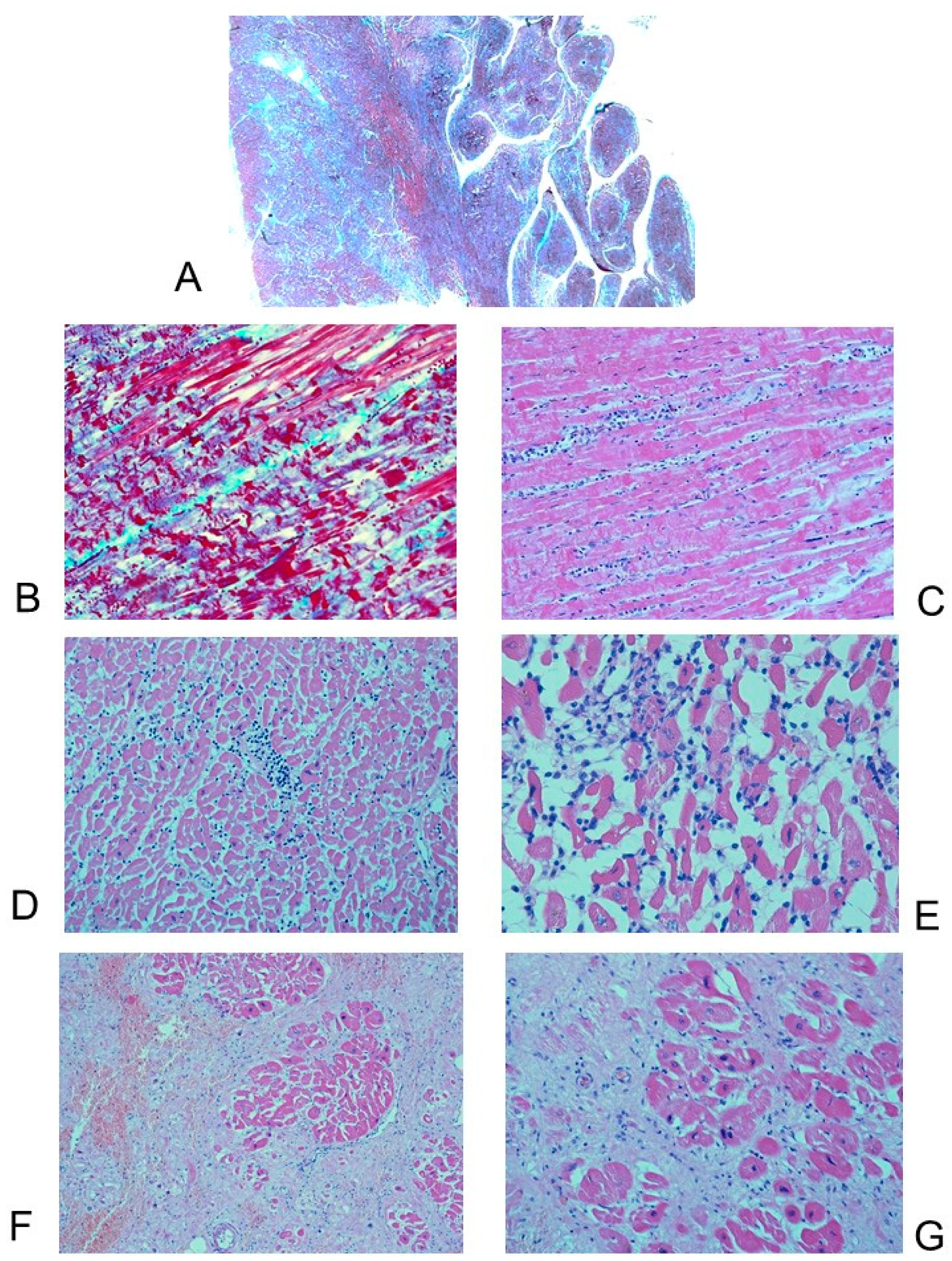
3.2.2. Microscopic Findings
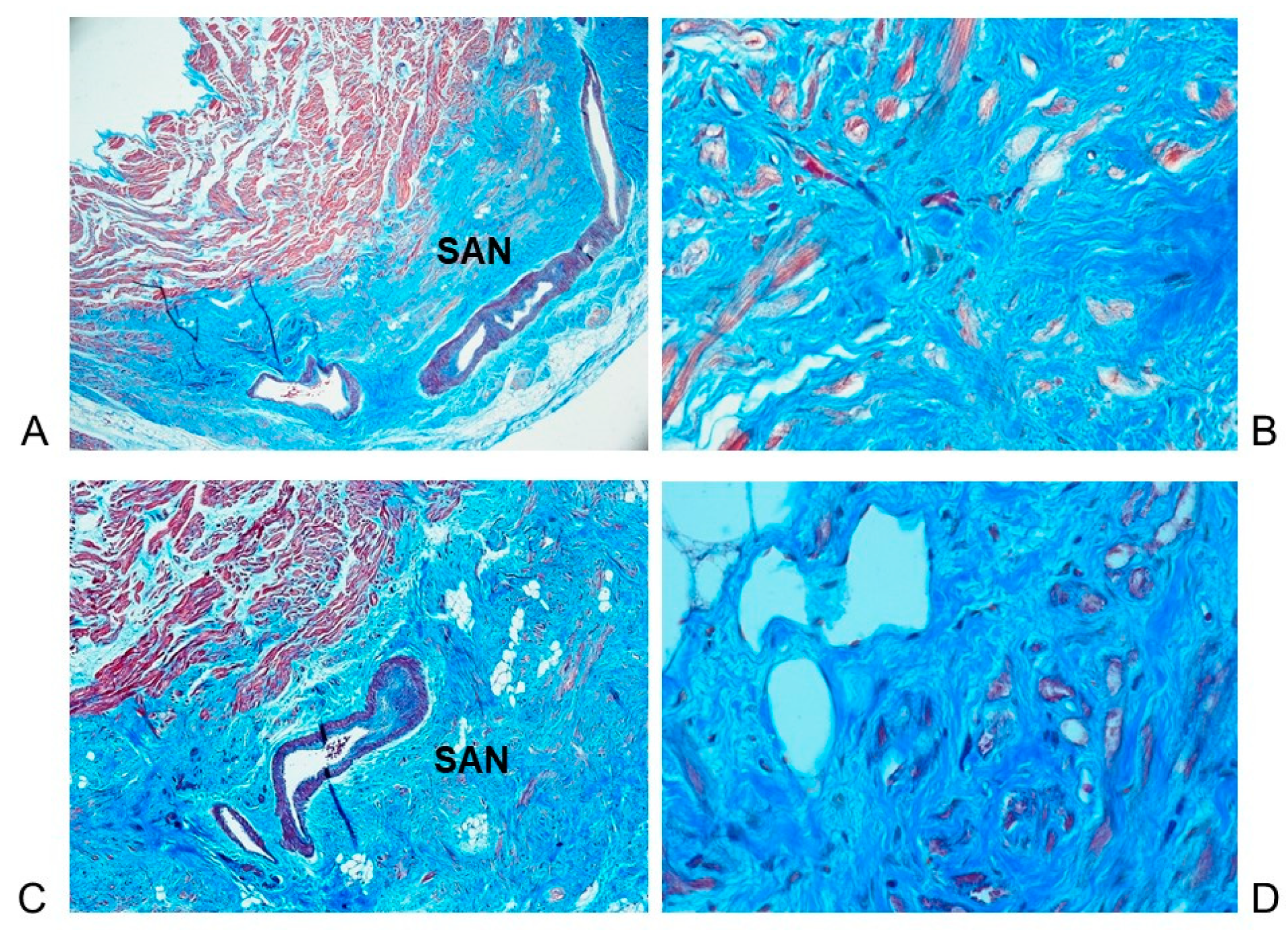
3.3. Histopathological Findings in the Cardiac Conduction System
4. Discussion
4.1. Patients’ Characteristics
4.2. Genetics
4.3. Anatomo-Pathological Findings of the Heart
4.4. Histopathological Findings in the Cardiac Conduction System
4.5. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Basso, C.; Judge, D. Arrhythmogenic cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.; Daubert, J.P.; De Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Hear. Rhythm. 2019, 16, e373–e407. [Google Scholar] [CrossRef]
- Ottaviani, G.; Buja, L.M. Anatomopathological changes of the cardiac conduction system in sudden cardiac death, particularly in infants: Advances over the last 25 years. Cardiovasc. Pathol. 2016, 25, 489–499. [Google Scholar] [CrossRef]
- Buja, L.M.; Ottaviani, G.; Mitchell, R.N. Pathobiology of cardiovascular diseases: An update. Cardiovasc. Pathol. 2019, 42, 44–53. [Google Scholar] [CrossRef]
- Thiene, G.; Corrado, M.; Basso, C. Sudden Cardiac Death in the Young and Athletes; Springer Science and Business Media LLC: Milan, Italy, 2016. [Google Scholar]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; McKenna, W.J.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2007, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic right ventricular cardiomyopathy: Characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J. Am. Hear. Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef] [PubMed]
- Matturri, L.; Ottaviani, G.; Lavezzi, A.M. Techniques and criteria in pathologic and forensic-medical diagnostics in sudden unexpected infant and perinatal death. Am. J. Clin. Pathol. 2005, 124, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Matturri, L.; Ottaviani, G.; Lavezzi, A. Guidelines for neuropathologic diagnostics of perinatal unexpected loss and sudden infant death syndrome (SIDS)—A technical protocol. Virchows Arch. 2007, 452, 19–25. [Google Scholar] [CrossRef]
- Ottaviani, G. Crib Death—Sudden Infant Death Syndrome (SIDS): Sudden Infant and Perinatal Unexplained Death: The Pathologist’s Viewpoint, 2nd ed.; Springer International Publishing: Heidelberg, Germany, 2014; ISBN 978-3-319-08346-9. [Google Scholar]
- Ottaviani, G.; Matturri, L. Histopathology of the cardiac conduction system in sudden intrauterine unexplained death. Cardiovasc. Pathol. 2008, 17, 146–155. [Google Scholar] [CrossRef]
- Rossi, L.; Matturri, L. Clinicopathological Approach to Cardiac Arrhythmias. A Color Atlas; Centro Scientifico Editore, Ed.; Futura Publishing Company: Turin, Italy; Mount Kisco, NY, USA, 1990. [Google Scholar]
- Matturri, L.; Ottaviani, G.; Ramos, S.G.; Rossi, L. Sudden Infant Death Syndrome (SIDS): A study of cardiac conduction system. Cardiovasc. Pathol. 2000, 9, 137–145. [Google Scholar] [CrossRef]
- Ottaviani, G.; Matturri, L.; Rossi, L.; James, T.N. Crib death: Further support for the concept of fatal cardiac electrical instability as the final common pathway. Int. J. Cardiol. 2003, 92, 17–26. [Google Scholar] [CrossRef]
- Matturri, L.; Ottaviani, G.; Lavezzi, A.M.; Rossi, L. Early atherosclerotic lesions of the cardiac conduction system arteries in infants. Cardiovasc. Pathol. 2004, 13, 276–281. [Google Scholar] [CrossRef]
- Matturri, L.; Ottaviani, G.; Rossi, L. Cardiac massage in infants. Intensive Care Med. 2003, 29, 1199–1200. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Nava, A.; Rossi, L.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia; Elsevier: Amsterdam, The Netherlands, 1997; ISBN 04441824472. [Google Scholar]
- Basso, C.; Pilichou, K.; Bauce, B.; Corrado, D.; Thiene, G. Diagnostic criteria, genetics, and molecular basis of arrhythmogenic cardiomyopathy. Heart Fail. Clin. 2018, 14, 201–213. [Google Scholar] [CrossRef]
- Tabib, A.; Loire, R.; Chalabreysse, L.; Meyronnet, D.; Miras, A.; Malicier, D.; Thivolet, F.; Chevalier, P.; Bouvagnet, P. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003, 108, 3000–3005. [Google Scholar] [CrossRef]
- Ottaviani, G.; Radovancevic, R.; Kar, B.; Gregoric, I.D.; Buja, L.M. Pathological assessment of end-stage heart failure in explanted hearts in correlation with hemodynamics in patients undergoing orthotopic heart transplantation. Cardiovasc. Pathol. 2015, 24, 283–289. [Google Scholar] [CrossRef]
- Roberts, W.C.; Roberts, C.C.; Ko, J.M.; Filardo, G.; Capehart, J.E.; Hall, S.A. Morphologic features of the recipient heart in patients having cardiac transplantation and analysis of the congruence or incongruence between the clinical and morphologic diagnoses. Medicine 2014, 93, 211–235. [Google Scholar] [CrossRef]
- Rakovec, P.; Rossi, L.; Fontaine, G.; Šašel, B.; Markež, J.; Vončina, D. Familial arrhythmogenic right ventricular disease. Am. J. Cardiol. 1986, 58, 377–378. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Moreau, A.; Reisqs, J.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessière, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 arrhythmogenic cardiomyopathy electrical instability: From ion channels to ECG and tailored drug therapy. Clin. Transl. Med. 2021, 11, e319. [Google Scholar] [CrossRef]
- Hayashi, K.; Teramoto, R.; Nomura, A.; Asano, Y.; Beerens, M.; Kurata, Y.; Kobayashi, I.; Fujino, N.; Furusho, H.; Sakata, K.; et al. Impact of functional studies on exome sequence variant interpretation in early-onset cardiac conduction system diseases. Cardiovasc. Res. 2020, 116, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Mattesi, G.; Zorzi, A.; Corrado, D.; Cipriani, A. Natural history of arrhythmogenic cardiomyopathy. J. Clin. Med. 2020, 9, 878. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy: Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.; Buja, L.M.; Kar, B.; Colnaric, J.; Damaraju, S.; Zhao, B.; Akkanti, B.; Radovanovic, M.; Radovancevic, R.; Loyalka, P.; et al. Cardiac sarcoidosis presenting as arrhythmogenic right ventricular cardiomyopathy/dysplasia with ventricular aneurysms: A case report. Cardiovasc. Pathol. 2018, 33, 1–5. [Google Scholar] [CrossRef]
- Buja, L.M.; Ottaviani, G.; Ilic, M.; Zhao, B.; Lelenwa, L.C.; Segura, A.M.; Bai, Y.; Chen, A.; Akkanti, B.; Hussain, R.; et al. Clinicopathological manifestations of myocarditis in a heart failure population. Cardiovasc. Pathol. 2020, 45, 107190. [Google Scholar] [CrossRef]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac fibro-adipocyte progenitors express Desmosome proteins and preferentially differentiate to adipocytes upon deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Russo, A.D.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Hear. J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef]
- Maione, A.; Stadiotti, I.; Pilato, C.; Perrucci, G.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-β1 drives cardiac mesenchymal stromal cells to a pro-fibrotic commitment in arrhythmogenic cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 2673. [Google Scholar] [CrossRef]
- Peters, S. Conduction abnormalities in arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2013, 168, 4920–4921. [Google Scholar] [CrossRef] [PubMed]
- Bharati, S.; Lev, M. Sudden death in athletes—Conduction system: Practical approach to dissection and pertinent pathology. Cardiovasc. Pathol. 1994, 3, 117–127. [Google Scholar] [CrossRef]
- Burke, A.; Virmani, R. Exercise as a factor in sudden cardiac death. Cardiovasc. Pathol. 1994, 3, 99–104. [Google Scholar] [CrossRef]
- Mezzano, V.; Pellman, J.; Sheikh, F. Cell junctions in the specialized conduction system of the heart. Cell Commun. Adhes. 2014, 21, 149–159. [Google Scholar] [CrossRef][Green Version]
- Corrado, D.; Basso, C.; Buja, G.; Nava, A.; Rossi, L.; Thiene, G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation 2001, 103, 710–717. [Google Scholar] [CrossRef]
- Grassi, S.; Vidal, M.; Campuzano, O.; Arena, V.; Alfonsetti, A.; Rossi, S.; Scarnicci, F.; Iglesias, A.; Brugada, R.; Oliva, A. Sudden Death without a clear cause after comprehensive investigation: An example of forensic approach to atypical/uncertain findings. Diagnostics 2021, 11, 886. [Google Scholar] [CrossRef]
- Matturri, L.; Ottaviani, G.; Lavezzi, A.; Turconi, P.; Cazzullo, A.; Rossi, L. Expression of apoptosis and proliferating cell nuclear antigen (PCNA) in the cardiac conduction system of crib death (SIDS). Adv. Clin. Path. 2001, 5, 79–86. [Google Scholar]
- James, T.N. Normal and abnormal consequences of apoptosis in the human heart. Annu. Rev. Physiol. 1998, 60, 309–325. [Google Scholar] [CrossRef]
- Kawashima, T.; Sato, F. First in situ 3D visualization of the human cardiac conduction system and its transformation associated with heart contour and inclination. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACM (N = 23) | |
|---|---|
| Gender (M/F) | 16 (69.56%)/7 (30.44%) |
| Mean age ± SD (years) | 36.13 ± 16.06; range: 5–65 |
| Ethnicity (W/B) | 22 (95.65%)/1 (4.35%) |
| Familial occurrence of SUCD < 50 years | 4 (17.4%) |
| Familial occurrence of ACM | 1 (4.35%) |
| Athlete | 1 (4.35%) |
| Previous symptoms | 5 (21.74%) |
| Diagnosis of ACM (IV/PM) | 3 (13.04%)/20 (86.96%) |
| ECG | 6 (26.08%) |
| Low QRS voltages | 2 (8.69%) |
| Epsilon waves | 1 (4.35%) |
| Incomplete right bundle block, Brugada syndrome | 1 (4.35%) |
| Ventricular fibrillation | 2 (8.69%) |
| Mode of death or failure: | |
| SUCD | 20 (86.96%) |
| at rest | 13 (56.52%) |
| on effort | 5 (21.74%) |
| unknown | 2 (8.69%) |
| CHF requiring OHT | 3 (13.04%) |
| ACM (N = 15) | |
|---|---|
| Gender (M/F) | 11 (77.33%)/4 (26.67%) |
| Mean age ± SD (years) | 35.07 ± 13.85; range: 5–51 |
| Ethnicity (W/B) | 14 (93.33%)/1 (6.67%) |
| Familial occurrence of SUCD < 50 years | 3 (20%) |
| Familial occurrence of ACM | 1 (6.67%) |
| AVJ hypoplasia due to fatty-fibrous involvement | 13 (86.67%) |
| SAN hypoplasia due to fatty-fibrous involvement | 12 (80%) |
| AVJ dispersion and/or septation | 7 (46.67%) |
| CFB hypoplasia | 5 (33.33%) |
| Fibromuscular Dysplasia of AVN artery | 3 (20%) |
| Fibromuscular Dysplasia of SAN artery | 2 (13.33%) |
| Islands of conduction tissue in CFB | 2 (13.33%) |
| Hemorrhage and infarct-like lesions in AVJ | 2 (13.33%) |
| LBB block by fibrosis | 2 (13.33%) |
| AVN tongue | 2 (13.33%) |
| Mahaim fiber | 2 (13.33%) |
| Hemorrhage in SAN | 1 (6.67%) |
| HB duplicity | 1 (6.67%) |
| Cartilaginous meta-hyperplasia of CFB | 1 (6.67%) |
| Right sided HB | 1 (6.67%) |
| N | Age (Years) | Sex | Heart Weight (g) | RV Bulging/Aneurysm | RV Wall Thickness (mm) | Patchy Inflammation | LV Involved | Cardiac Conduction System |
|---|---|---|---|---|---|---|---|---|
| 1 | 17 | M | 430 | Yes | 4 | No | Yes | SAN and AVJ fatty-fibrous involvement; right sided HB |
| 2 | 17 | M | 510 | No | 5 | No | No | SAN and AVJ fatty-fibrous involvement; AVJ septation |
| 3 | 29 | M | 408 | No | 5 | No | No | SAN and AVJ fatty-fibrous involvement; islands of conduction tissue in CFB; hemorrhage and infarct-like lesions in AVJ; LBB block by fibrosis |
| 4 | 40 | M | 508 | No | 4.5 | No | No | SAN and AVJ fatty-fibrous involvement |
| 5 | 33 | M | 500 | Yes | 2 | No | Yes | SAN and AVJ fatty-fibrous involvement; AVJ dispersion |
| 6 | 5 | F | 92 | Yes | 2 | Yes | No | SAN fatty-fibrous involvement; septated HB; CFB hypoplasia; Mahaim fiber LBB-ventricular (lower type); Cartilaginous meta-hyperplasia of CFB |
| 7 | 44 | M | 664 | Yes | 5 | Yes | Yes | SAN and AVJ fatty-fibrous involvement; septated HB; CFB hypoplasia |
| 8 | 46 | F | 377 | No | 8 | No | Yes | AVJ fatty-fibrous involvement; AVJ dispersion; SAN artery fibromuscular dysplasia |
| 9 | 47 | F | 371 | Yes | 5 | No | Yes | Hemorrhage in SAN; AVJ fatty-fibrous involvement; fibromuscular dysplasia of AVN artery; Mahaim fiber fasciculo-ventricular (middle type); |
| 10 | 31 | M | 419 | Yes | 3 | No | Yes | SAN and AVJ fatty-fibrous involvement; septated AVN and HB; CFB hypoplasia; fibromuscular dysplasia of AVN artery; islands of conduction tissue in CFB; HB duplicity |
| 11 | 51 | F | 288 | No | 8 | No | Yes | SAN and AVJ fatty-fibrous involvement; AVJ dispersion; LBB block by fibrosis |
| 12 | 41 | M | 391 | Yes | 5 | No | Yes | SAN and AVJ fatty-fibrous involvement; fibromuscular dysplasia of AVN artery |
| 13 | 47 | M | 415 | Yes | 5 | Yes | No | SAN and AVJ fatty-fibrous involvement; hemorrhage and infarct-like lesions in AVJ |
| 14 | 50 | M | 440 | Yes | 6 | No | No | SAN and AVJ fatty-fibrous involvement; CFB hypoplasia; SAN artery fibromuscular dysplasia |
| 15 | 28 | M | 450 | Yes | 4 | No | No | Unremarkable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviani, G.; Alfonsi, G.; Ramos, S.G.; Buja, L.M. Sudden Unexpected Death Associated with Arrhythmogenic Cardiomyopathy: Study of the Cardiac Conduction System. Diagnostics 2021, 11, 1323. https://doi.org/10.3390/diagnostics11081323
Ottaviani G, Alfonsi G, Ramos SG, Buja LM. Sudden Unexpected Death Associated with Arrhythmogenic Cardiomyopathy: Study of the Cardiac Conduction System. Diagnostics. 2021; 11(8):1323. https://doi.org/10.3390/diagnostics11081323
Chicago/Turabian StyleOttaviani, Giulia, Graziella Alfonsi, Simone G. Ramos, and L. Maximilian Buja. 2021. "Sudden Unexpected Death Associated with Arrhythmogenic Cardiomyopathy: Study of the Cardiac Conduction System" Diagnostics 11, no. 8: 1323. https://doi.org/10.3390/diagnostics11081323
APA StyleOttaviani, G., Alfonsi, G., Ramos, S. G., & Buja, L. M. (2021). Sudden Unexpected Death Associated with Arrhythmogenic Cardiomyopathy: Study of the Cardiac Conduction System. Diagnostics, 11(8), 1323. https://doi.org/10.3390/diagnostics11081323