
A Rare Case of Allantoic Cyst with Patent Urachus in Fetus with a Microdeletion in 1q21.1q21.2 Region


 and
and 
{kind=link}
{kind=link}
Abstract
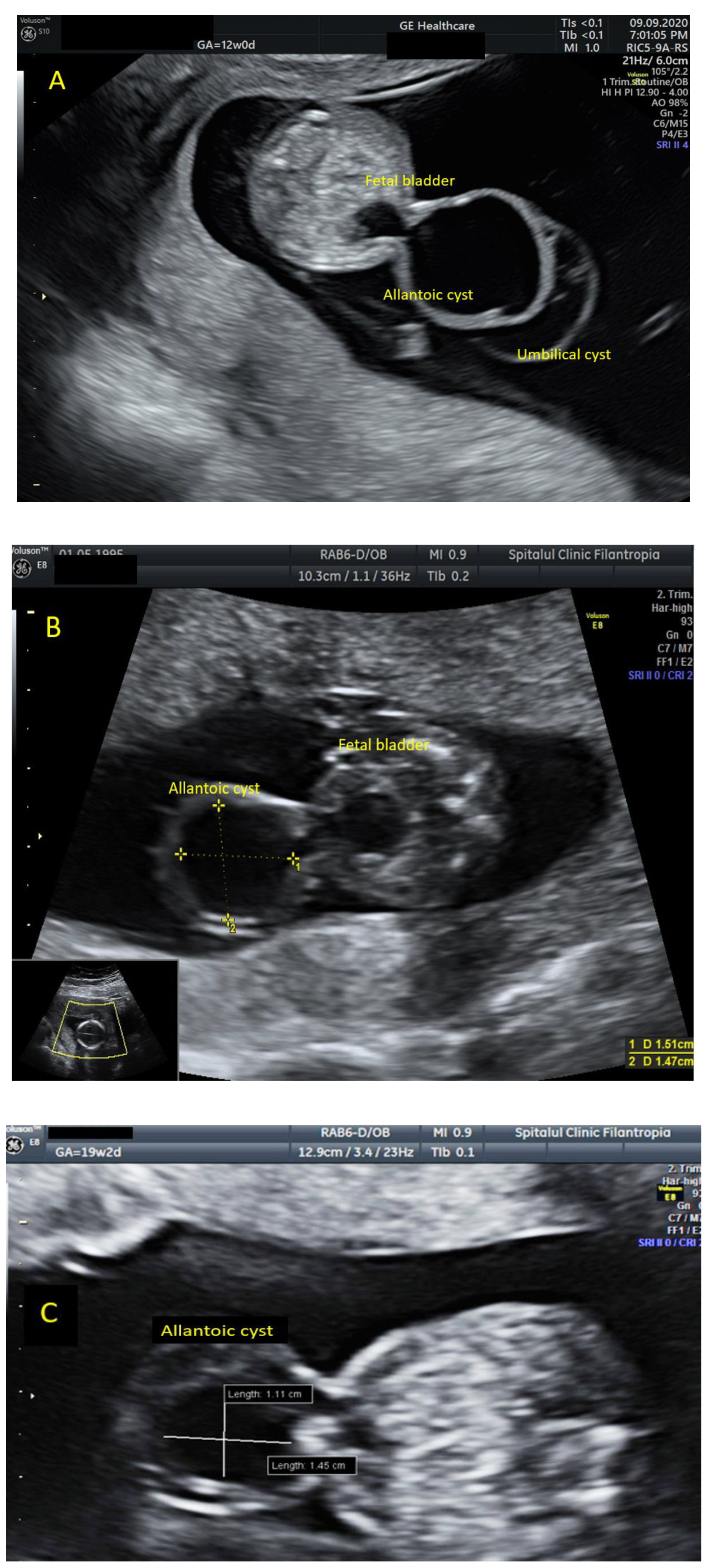
:
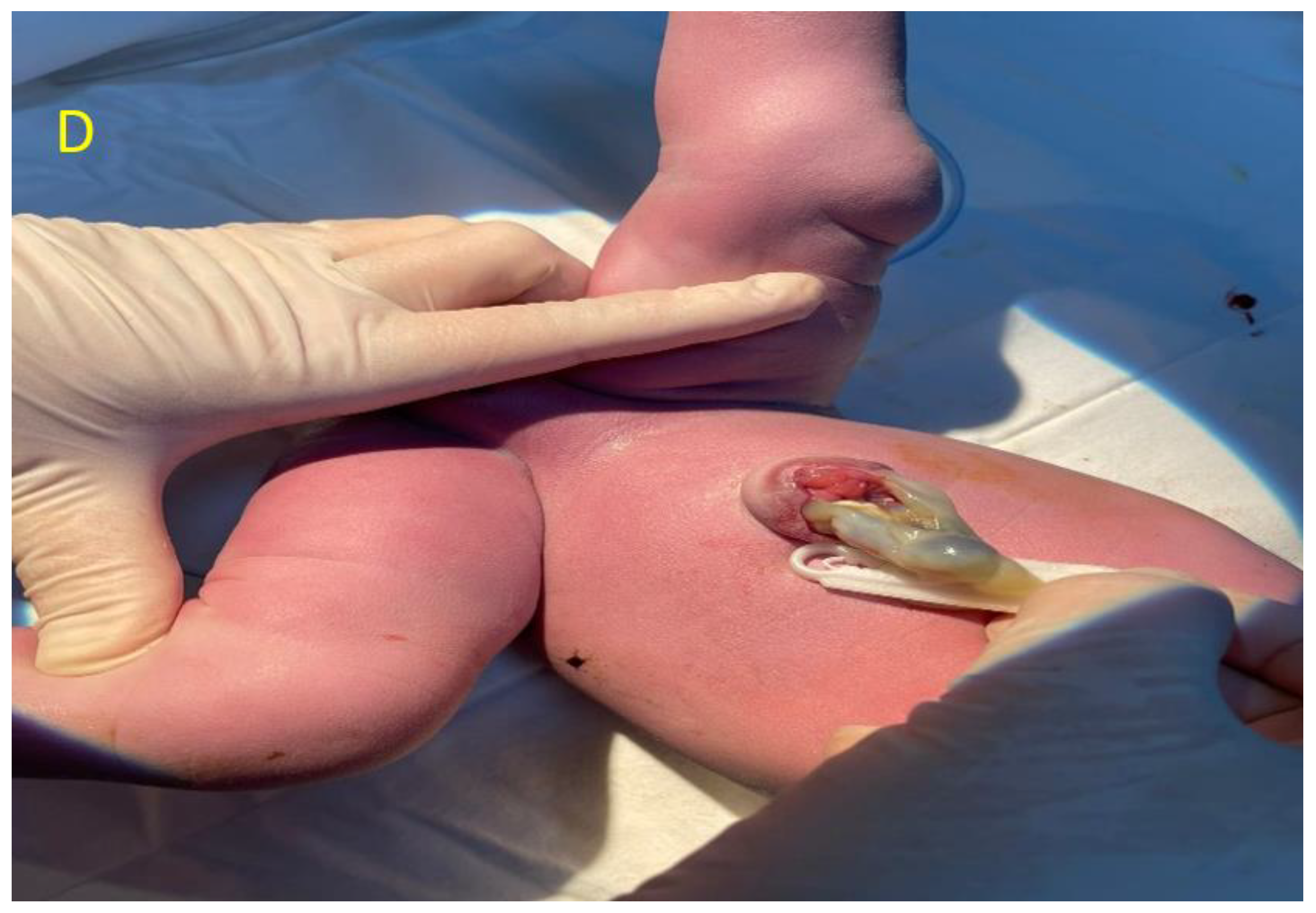
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Veduta, A.; Vayna, A.M.; Duta, S.; Panaitescu, A.; Popescu, F.; Bari, M.; Peltecu, G.; Nedelea, F. The first trimester combined test for aneuploidies—A single center experience. J. Matern. Neonatal Med. 2017, 31, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Vayna, A.M.; Veduta, A.; Duta, S.; Panaitescu, A.M.; Stoica, S.; Buinoiu, N.; Nedelea, F.; Peltecu, G. Diagnosis of Fetal Structural Anomalies at 11 to 14 Weeks. J. Ultrasound Med. 2018, 37, 2063–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, P.; Retik, A.; Vaughan, E.D., Jr.; Wein, A.J.; Kavoussi, L.R.; Novick, A.C. Other bladder anomalies. In Campbell’s Urology, 8th ed.; Saunders: Philadelphia, PA, USA, 2002; pp. 2189–2191. [Google Scholar]
- Yu, J.-S.; Kim, K.W.; Lee, H.-J.; Lee, Y.-J.; Yoon, C.-S.; Kim, M.-J. Urachal remnant disease: Spectrum of CT and US findings. Radiographics 2001, 21, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, S.; Usui, N.; Kanagawa, T.; Yamamichi, T.; Nara, K.; Ueno, T.; Owari, M.; Uehara, S.; Oue, T.; Kimura, T.; et al. Prenatal and Postnatal Clinical Course of an Urachus Identified as an Allantoic Cyst in the Umbilical Cord. Eur. J. Pediatr. Surg. 2016, 26, 200–202. [Google Scholar] [PubMed]
- Haldeman-Englert, C.R.; Jewett, T. 1q21.1 Recurrent Microdeletion. GeneReviews 2015, 8, 93–97. [Google Scholar]
- Mefford, H.; Hulten, M. 1q21.1 microdeletions. Unique 2014, 16, 359. [Google Scholar]
- Riddell, J.V.B.; Houle, A.; Franc-Guimond, J.; Barrieras, D. Prenatal vesico-allantoic cyst outcome—A spectrum from patent urachus to bladder exstrophy. Prenat. Diagn. 2015, 35, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Morichon-Delvallez, N. 1q21.1 Microdeletion Syndrome. March 2011. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=250989 (accessed on 26 October 2021).
- Harvard, C.; Strong, E.; Mercier, E.; Colnaghi, R.; Alcantara, D.; Chow, E.; Martell, S.; Tyson, C.; Hrynchak, M.; McGillivray, B.; et al. Understanding the impact of 1q21.1 copy number variant. Orphanet J. Rare Dis. 2011, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouariu, A.; Scutelnicu, A.M.; Ciobanu, A.M.; Cimpoca Raptis, B.A.; Dumitru, A.E.; Nedelea, F.; Gică, N.; Panaitescu, A.M. A Rare Case of Allantoic Cyst with Patent Urachus in Fetus with a Microdeletion in 1q21.1q21.2 Region. Diagnostics 2021, 11, 2332. https://doi.org/10.3390/diagnostics11122332
Bouariu A, Scutelnicu AM, Ciobanu AM, Cimpoca Raptis BA, Dumitru AE, Nedelea F, Gică N, Panaitescu AM. A Rare Case of Allantoic Cyst with Patent Urachus in Fetus with a Microdeletion in 1q21.1q21.2 Region. Diagnostics. 2021; 11(12):2332. https://doi.org/10.3390/diagnostics11122332
Chicago/Turabian StyleBouariu, Alexandra, Ana Maria Scutelnicu, Anca Marina Ciobanu, Brîndușa Ana Cimpoca Raptis, Andreea Elena Dumitru, Florina Nedelea, Nicolae Gică, and Anca Maria Panaitescu. 2021. "A Rare Case of Allantoic Cyst with Patent Urachus in Fetus with a Microdeletion in 1q21.1q21.2 Region" Diagnostics 11, no. 12: 2332. https://doi.org/10.3390/diagnostics11122332
APA StyleBouariu, A., Scutelnicu, A. M., Ciobanu, A. M., Cimpoca Raptis, B. A., Dumitru, A. E., Nedelea, F., Gică, N., & Panaitescu, A. M. (2021). A Rare Case of Allantoic Cyst with Patent Urachus in Fetus with a Microdeletion in 1q21.1q21.2 Region. Diagnostics, 11(12), 2332. https://doi.org/10.3390/diagnostics11122332