
Low Represented Mutation Clustering in SARS-CoV-2 B.1.1.7 Sublineage Group with Synonymous Mutations in the E Gene
 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
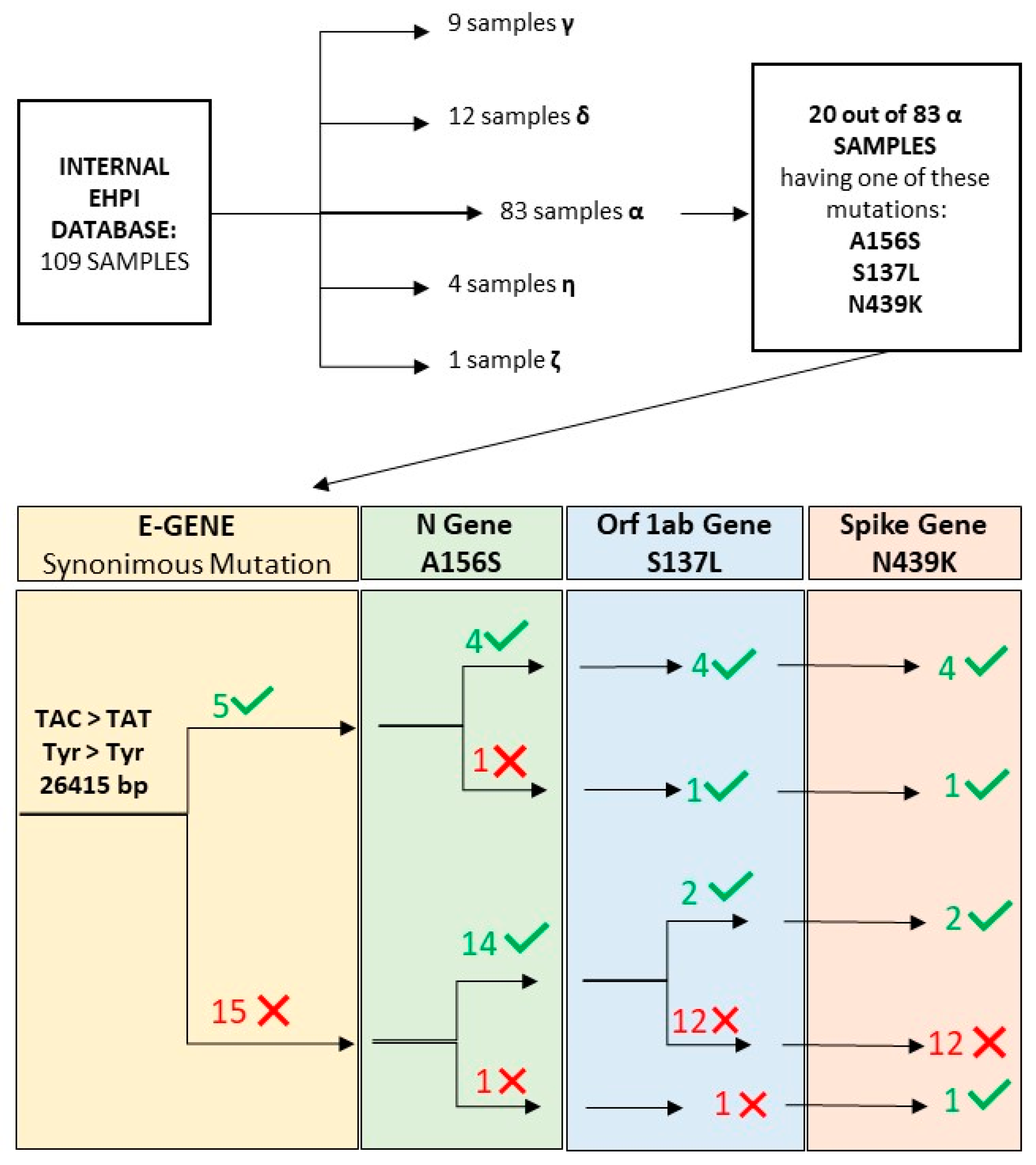
2.1. Samples Included in the Study
2.2. Internal Database
2.3. Primer Design, RNA Reverse Transcription and PCR Amplification
2.4. Purification of PCR Products, SANGER Sequencing and Sequence Analysis
2.5. Next Generation Sequencing (NGS) on the MiSeq Illumina® Platform
2.6. Bioinformatic Analysis
3. Results
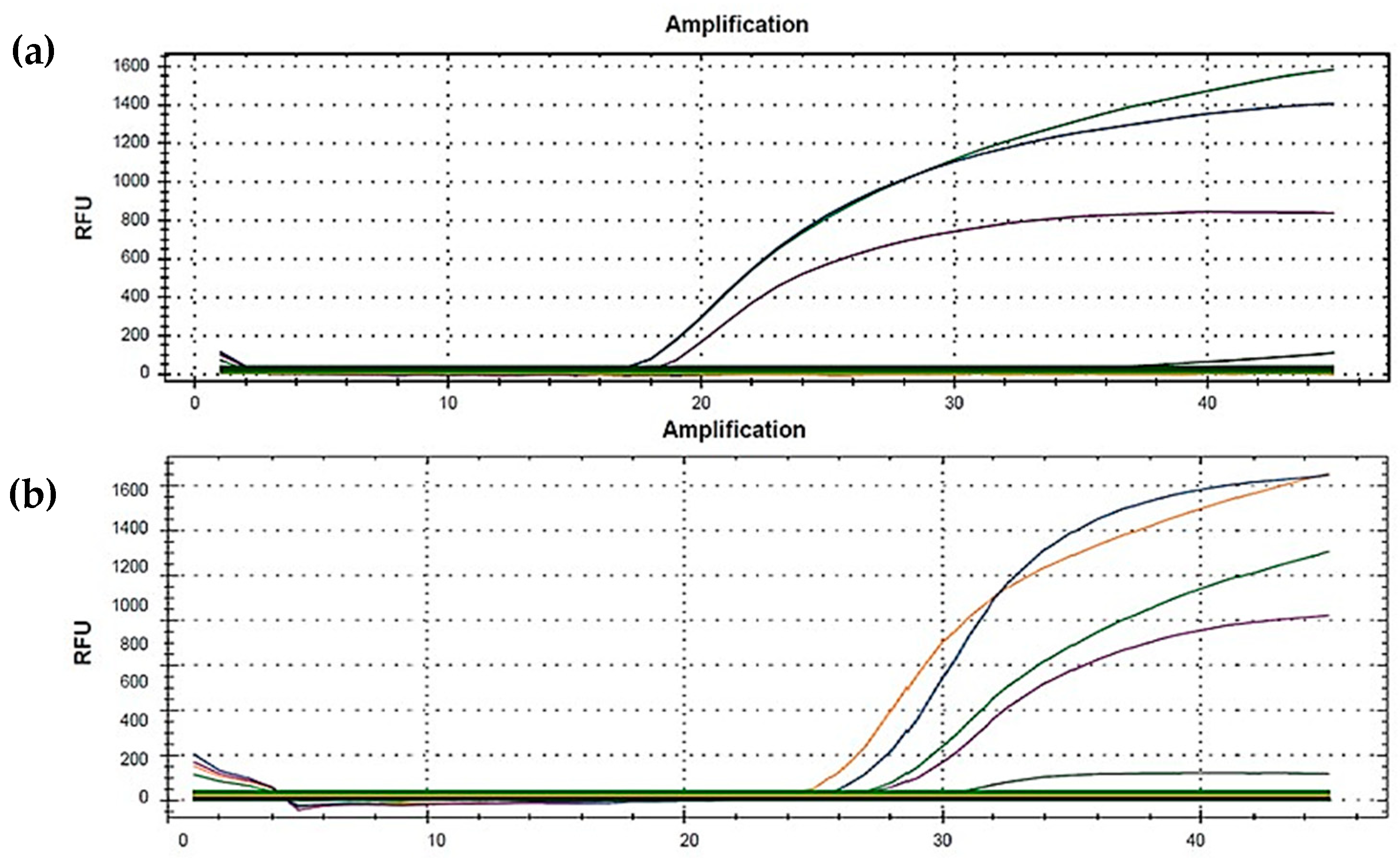
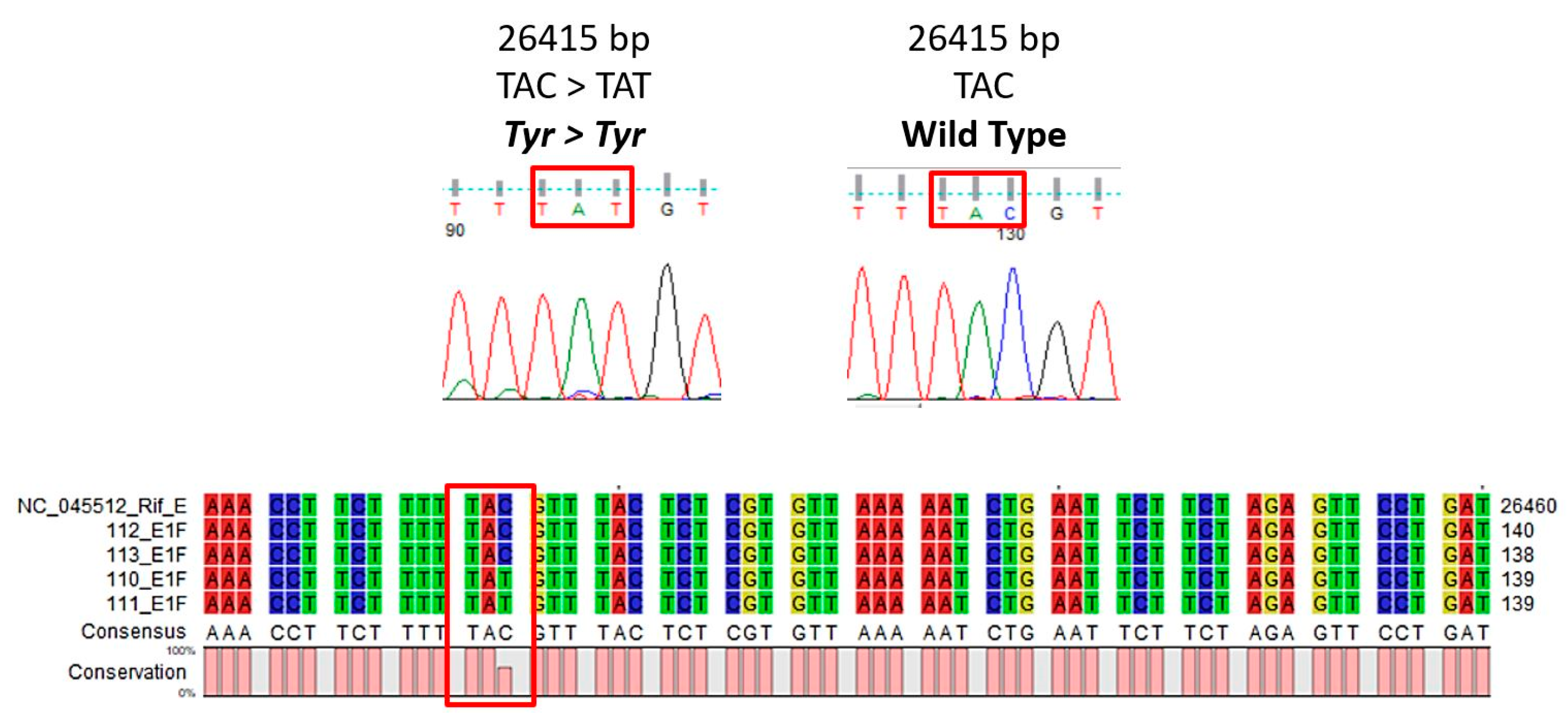
3.1. E Gene Mutation Analysis
3.2. NGS Sequencing Analysis
3.3. Statistical Analysis of Mutation Frequency
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, H.; Gupte, J.; Fletcher, H.; Hammond, L.; Lowe, N.; Pelling, M.; Raina, N.; Shahid, T.; Shanks, K. COVID-19 as a Global Challenge: Towards an Inclusive and Sustainable Future; Elsevier Ltd.: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 3 November 2021).
- European Centre for Disease Prevention and Control. Sequencing of SARS-CoV-2: First Update; ECDC: Stockholm, Sweden, 2021.
- Maurier, F.; Beury, D.; Fléchon, L.; Varré, J.S.; Touzet, H.; Goffard, A.; Hot, D.; Caboche, S. A complete protocol for whole-genome sequencing of virus from clinical samples: Application to coronavirus OC43. Virology 2019, 531, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Capoferri, A.A.; Katusiime, M.G.; Patro, S.C.; Kearney, M.F. Low genetic diversity may be an Achilles heel of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 24614–24616. [Google Scholar] [CrossRef]
- Jogalekar, M.P.; Veerabathini, A.; Gangadaran, P. SARS-CoV-2 variants: A double-edged sword? Exp. Biol. Med. 2021, 246, 1721–1726. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. SARS-CoV-2 mutations: The biological trackway towards viral fitness. Epidemiol. Infect. 2021, 149, E110. [Google Scholar] [CrossRef]
- Singh, D.; Soojin, V.Y. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Nguyen PT, T.; Plyusnin, I.; Sironen, T.; Vapalathi, O.; Kant, R.; Smura, T. HAVoC, a bioinformatic pipeline for reference-based consensus assembly and lineage assignment for SARS-CoV-2 sequences. BMC Bioinform. 2021, 22, 373. [Google Scholar]
- Kalia, K.; Saberwal, G.; Sharma, G. The lag in SARS-CoV-2 genome submissions to GISAID. Nat. Biotechnol. 2021, 39, 1058–1060. [Google Scholar] [CrossRef]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; Yeats, C.; et al. Pangolin: Lineage Assignment in an Emerging Pandemic as an Epidemiological Tool. Available online: https://github.com/cov-lineages/pangolin (accessed on 3 November 2021).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Yuelong, S.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef] [PubMed]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Wollschlager, P.; Todt, D.; Gerlitz, N.; Pfaender, S.; Bollinger, T.; Sing, A.; Dangel, A.; Ackermann, N.; Korn, K.; Ensser, A.; et al. SARS-CoV-2 N gene dropout and N gene Ct value shift as indicator for the presence of B.1.1.7 lineage in a commercial multiplex PCR assay. Clin. Microbiol. Infect. 2021, 27, 1353.E1–1353.E5. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Eurosurveillance 2020, 25, 2001650. [Google Scholar] [CrossRef]
- Artesi, M.; Bontems, S.; Gobbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.P.; Bours, V.; et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. ASM J. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Musso, N.; Falzone, L.; Stracquadanio, S.; Bongiorno, D.; Salerno, M.; Esposito, M.; Sessa, F.; Libra, M.; Pomara, C. Post-Mortem Detection of SARS-CoV-2 RNA in Long-Buried Lung Samples. Diagnostics 2021, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Musso, N.; Costantino, A.; La Spina, S.; Finocchiaro, A.; Andronico, F.; Stracquadanio, S.; Liotta, L.; Visalli, R.; Emmanuele, G. New SARS-CoV-2 Infection Detected in an Italian Pet Cat by RT-qPCR from Deep Pharyngeal Swab. Pathogens 2020, 9, 746. [Google Scholar] [CrossRef]
- Falzone, L.; Musso, N.; Gattuso, G.; Bongiorno, D.; Palermo, C.I.; Scalia, G.; Libra, M.; Stefani, S. Sensitivity assessment of droplet digital PCR for SARS-CoV-2 detection. Int. J. Mol. Med. 2020, 46, 957–964. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization: Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 3 November 2021).
- Colson, P.; Raoult, D. Global Discrepancies between Numbers of Available SARS-CoV-2 Genomes and Human Development Indexes at Country Scales. Viruses 2021, 13, 775. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Martinez, I.L.; Llinàs, D.T.; Romero, M.B.; Salazar, L.M. High Mutation Rate in SARS-CoV-2: Will It Hit Us the Same Way Forever? J. Infect. Dis. Epidemiol. 2020, 6, 176. [Google Scholar]
- Lo Presti, A.; Rezza, G.; Stefanelli, P. Selective pressure on SARS-CoV-2 protein coding genes and glycosylation site prediction. Heliyon 2020, 6, e05001. [Google Scholar] [CrossRef] [PubMed]
- GISAID: Submission Tracker Global. Available online: https://www.gisaid.org/index.php?id=208 (accessed on 3 November 2021).
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.A.; Akinduti, P.A.; Arege, O.T.; Obafemi, Y.D.; Faneye, A.O.; Isibor, P.O.; Aworunse, O.S.; Oranusi, S.U. Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int. J. Infect. Dis. 2021, 103, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Tramuto, F.; Reale, S.; Lo Presti, A.; Vitale, F.; Pulvirenti, C.; Rezza, G.; Vitale, F.; Purpari, G.; Maida, C.M.; Zichichi, S.; et al. Genomic Analysis and Lineage Identification of SARS-CoV-2 Strains in Migrants Accessing Europe Through the Libyan Route. Front. Public Health 2021, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Deckert, A.; Anders, S.; De Allegri, M.; Nguyen, H.T.; Souares, A.; McMahon, S.; Meurer, M.; Burk, R.; Sand, M.; Koeppel, L.; et al. Effectiveness and cost-effectiveness of four different strategies for SARS-CoV-2 surveillance in the general population (CoV-Surv Study): Study protocol for a two-factorial randomized controlled multi-arm trial with cluster sampling. Trials 2021, 22, 656. [Google Scholar] [CrossRef] [PubMed]
- Quéromes, G.; Destras, G.; Bal, A.; Regue, H.; Burfin, G.; Brun, S.; Fanget, R.; Morfin, F.; Valette, M.; Trouillet-Assant, S.; et al. Characterization of SARS-CoV-2 ORF6 deletion variants detected in a nosocomial cluster during routine genomic surveillance, Lyon, France. Emerg. Microbes Infect. 2021, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Patient | Average Coverage | c.26415 Coverage | A156S Coverage | N439K Coverage | S139L Coverage | Quality Score (Probability of Incorrect Base Call) |
|---|---|---|---|---|---|---|
| 1 | 5505 | 2930 | 14,584 | 7345 | 4105 | 95% > Q30 (1 in 1.000) |
| 2 | 5390 | 3025 | 13,304 | 8207 | 4625 | |
| 3 | 16,937 | 3125 | 75,924 | 2366 | 24,526 | |
| 4 | 13,607 | 1301 | 51,036 | 2505 | 26,154 | |
| 5 | 4732 | 194 | - | 7160 | 826 |
| Gene | Primer Sequences 5′-3′ | Position in Reference Genome | Amplicon Size (bp) | |
|---|---|---|---|---|
| E gene | 329 | |||
| E1 | F | TCTTTTTCTTGCTTTCGTGGT | 26,298 | |
| R | ACAACCGTATCCGTTTAACA | 26,627 | ||
| S gene | ||||
| FS-4 | F | CCGCATCATTTTCCACTTTT | 22,674 | 825 |
| R | CACGTCCGACAAATTATCCCC | 23,499 | ||
| FS-5 | F | TTTGGTTAAAAACAAATGTGTCAA | 23,158 | 828 |
| R | AGGTAGTTTTGGTTCGTTCT | 23,986 | ||
| Mutation | Gene | GISAID Function | GISAID Frequency | EHPI Frequency | World Frequency |
|---|---|---|---|---|---|
| S137L | Orf 1ab | Not reported | 0.12% | 6.42% | 0.21% |
| N439K | Spike | Surface Receptor Binding Antibody Recognition Sites—Viral Oligomerization Interfaces-Antigenic Drift | 1.43% | 7.33% | 1.07% |
| A156S | Nucleocapsid | Viral Oligomerization Interface—Ligand Binding—Antigenic Drift | 0.25% | 21.20% | 0.21% |
| EHPI vs. Italy | EHPI vs. World | Italy vs. World | |
|---|---|---|---|
| S137L | 0.290604089 | 1 × 107—126 **** | 2.38 × 10—11 **** |
| N439K | 0.000146076 *** | 8.99 × 10—10 **** | 2.00 × 10—81 **** |
| A156S | 3.95 × 10—53 **** | 0 **** | 0 **** |
| E gene c.26415 C > T vs. Wild Type | |||
| S137L | 0.000433736 *** | ||
| N439K | 0.001565402 ** | ||
| A156S | 0.389423696 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonacci, P.G.; Bivona, D.A.; Bongiorno, D.; Stracquadanio, S.; Massimino, M.; Bonomo, C.; Stracuzzi, A.; Pennisi, P.; Musso, N.; Stefani, S. Low Represented Mutation Clustering in SARS-CoV-2 B.1.1.7 Sublineage Group with Synonymous Mutations in the E Gene. Diagnostics 2021, 11, 2286. https://doi.org/10.3390/diagnostics11122286
Bonacci PG, Bivona DA, Bongiorno D, Stracquadanio S, Massimino M, Bonomo C, Stracuzzi A, Pennisi P, Musso N, Stefani S. Low Represented Mutation Clustering in SARS-CoV-2 B.1.1.7 Sublineage Group with Synonymous Mutations in the E Gene. Diagnostics. 2021; 11(12):2286. https://doi.org/10.3390/diagnostics11122286
Chicago/Turabian StyleBonacci, Paolo Giuseppe, Dalida Angela Bivona, Dafne Bongiorno, Stefano Stracquadanio, Mariacristina Massimino, Carmelo Bonomo, Alessia Stracuzzi, Paolo Pennisi, Nicolò Musso, and Stefania Stefani. 2021. "Low Represented Mutation Clustering in SARS-CoV-2 B.1.1.7 Sublineage Group with Synonymous Mutations in the E Gene" Diagnostics 11, no. 12: 2286. https://doi.org/10.3390/diagnostics11122286
APA StyleBonacci, P. G., Bivona, D. A., Bongiorno, D., Stracquadanio, S., Massimino, M., Bonomo, C., Stracuzzi, A., Pennisi, P., Musso, N., & Stefani, S. (2021). Low Represented Mutation Clustering in SARS-CoV-2 B.1.1.7 Sublineage Group with Synonymous Mutations in the E Gene. Diagnostics, 11(12), 2286. https://doi.org/10.3390/diagnostics11122286