Metabolic Endotoxemia, Feeding Studies and the Use of the Limulus Amebocyte (LAL) Assay; Is It Fit for Purpose?


Abstract
1. Introduction
2. Materials and Methods
2.1. Participant Recruitment
2.2. Anthropometric Measurements
2.3. Blood Collection
2.4. Assessment of Metabolic Endotoxemia
2.4.1. Limulus Amebocyte (LAL) Assay
Preparation of Standards
Preparations of Plasma Samples
Preparation of Plasma Samples; Adaption for Ultrasonication
Preparation of Plasma Samples; Adaption for Addition of Pyrosperse
LAL Analysis
Summary of Polynomial Model Validation and Quality Measures
2.4.2. Lipopolysaccharide Binding Protein (LBP) Endotoxin Measurement
2.5. Data Interpretation
3. Results
3.1. Participant Demographics
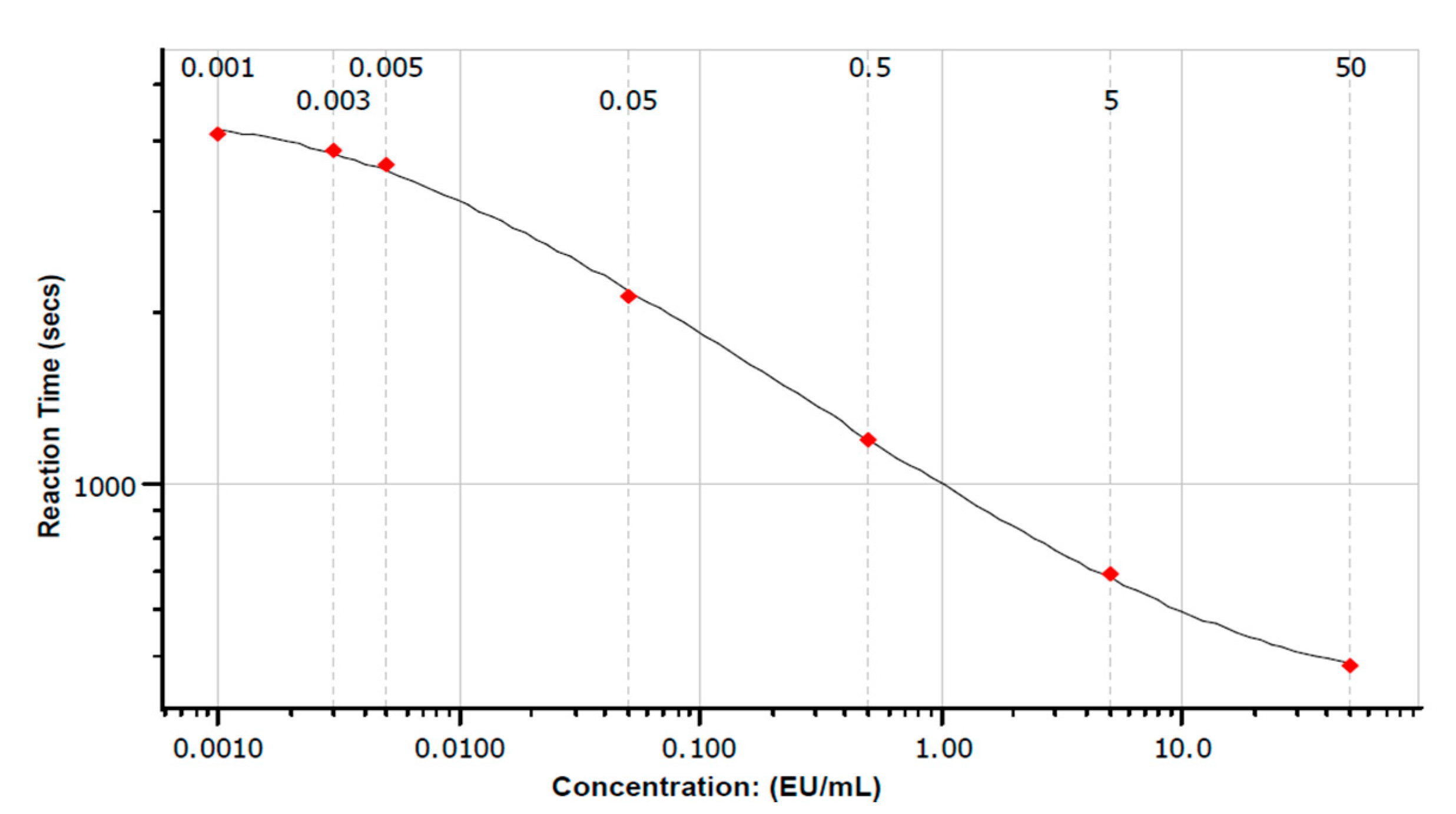
3.2. LAL Analysis
3.2.1. Summary of Polynomial Model Validation and Quality Measures
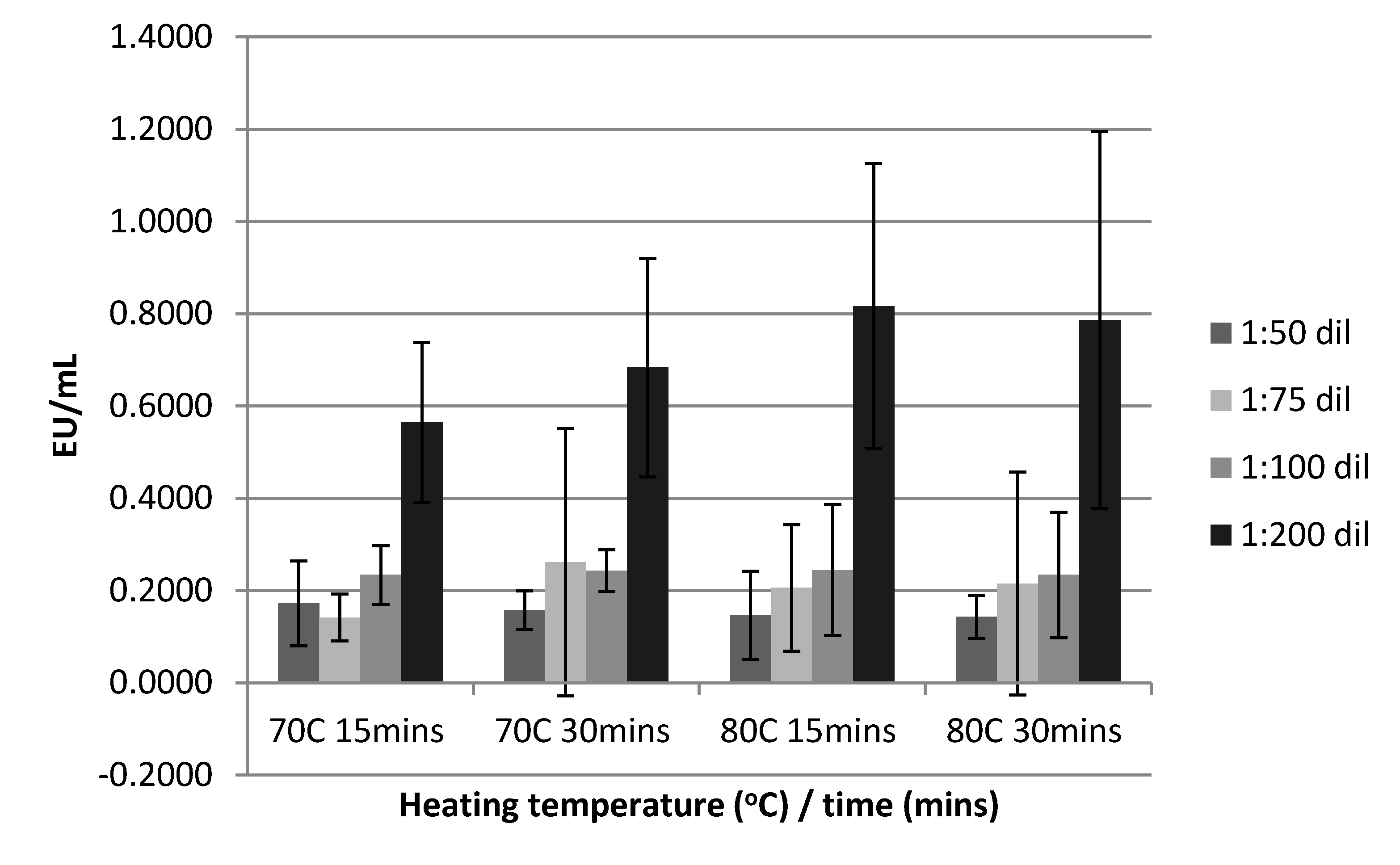
3.2.2. Sample Heat Treatment, Deactivation Time and Dilution on Serum Endotoxin Levels
3.2.3. LAL Adaption for Ultrasonication
3.2.4. LAL Adaption for Addition of Pyrosperse
3.2.5. Relationship between BMI and Endotoxemia
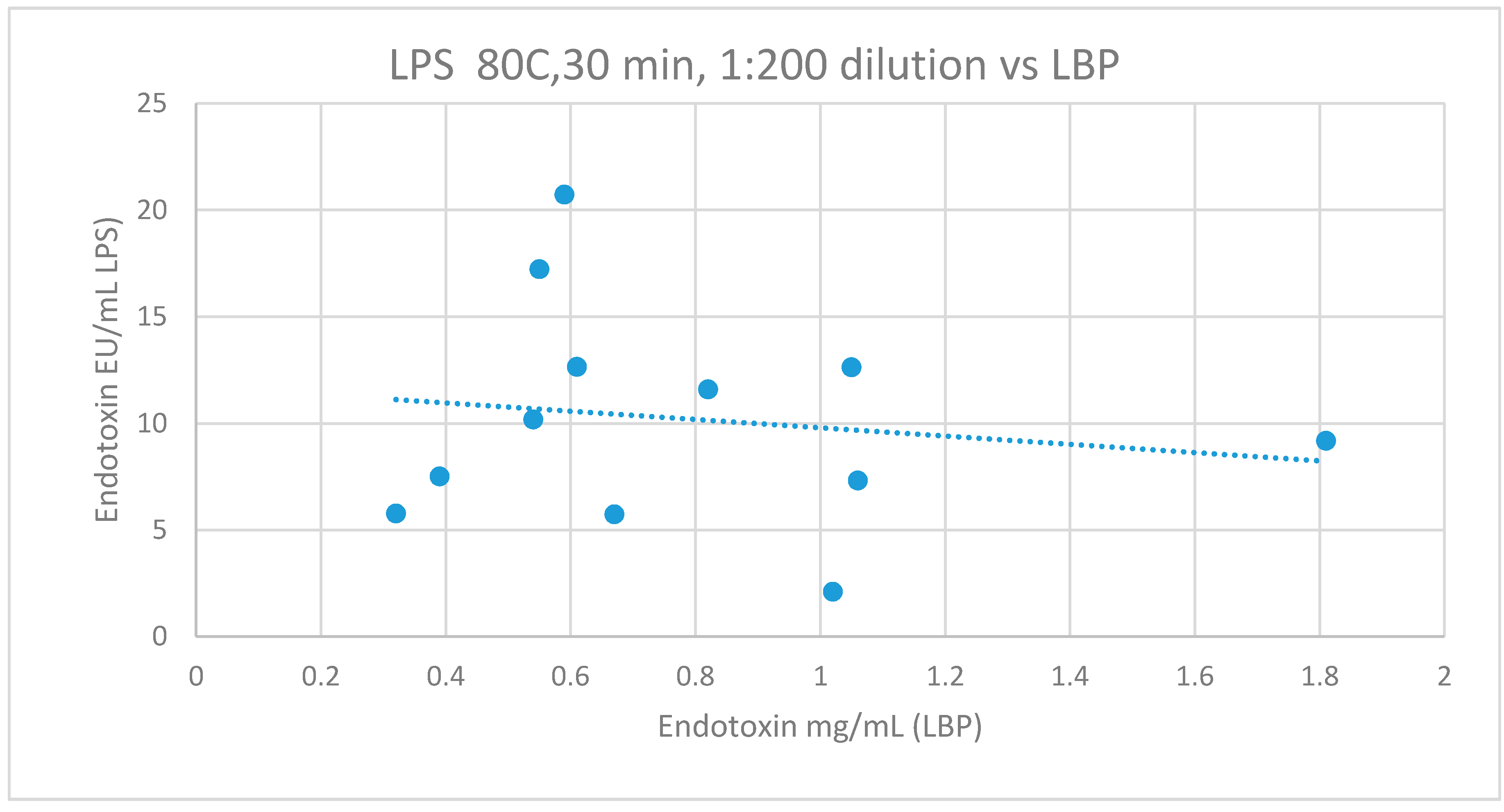
3.2.6. Comparison of LBP to LAL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51. [Google Scholar] [CrossRef] [PubMed]
- Wiedermann, C.J.; Kiechl, S.; Dunzendorfer, S.; Schratzberger, P.; Egger, G.; Oberhollenzer, F.; Willeit, J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the bruneck study. J. Am. Coll. Cardiol. 1999, 34, 1975–1981. [Google Scholar] [CrossRef]
- Harte, A.L.; Da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef]
- McPhee, N.; Tremellen, K.; Pearce, K.L. Evaluation of the use of the Endotoxin Activity Assay (EAA™) to Quantify Non-septic Exposure of Metabolic Endotoxemia. J. Med. Diagn. Methods 2017, 6, 4. [Google Scholar] [CrossRef]
- Schumann, R.R.; Zweigner, J. A novel acute-phase marker: Lipopolysaccharide binding protein (LBP). Clin. Chem. Lab. Med. 1999, 37, 271–274. [Google Scholar] [CrossRef]
- Weiss, J. Bactericidal/Permeability-Increasing Protein (BPI) and Lipopolysaccharide-Binding Protein (LBP): Structure, Function and Regulation in Host Defence against Gram-Negative Bacteria. Biochem. Soc. Trans. 2003, 4, 785–790. [Google Scholar] [CrossRef]
- Sun, L.; Yu, Z.; Ye, X.; Zou, S.; Li, H.; Yu, D.; Wu, H.; Chen, Y.; Dore, J.; Clément, K. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care 2010, 33, 1925–1932. [Google Scholar] [CrossRef]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Gonzalez-Quintela, A.; Alonso, M.; Campos, J.; Vizcaino, L.; Loidi, L.; Gude, F. Determinants of serum concentrations of lipopolysaccharide-binding protein (LBP) in the adult population: The role of obesity. PLoS ONE 2013, 8, e54600. [Google Scholar] [CrossRef]
- Liu, X.; Lu, L.; Yao, P.; Ma, Y.; Wang, F.; Jin, Q.; Ye, X.; Li, H.; Hu, F.B.; Sun, L.; et al. Lipopolysaccharide binding protein, obesity status and incidence of metabolic syndrome: A prospective study among middle-aged and older Chinese. Diabetologia 2014, 57, 1834–1841. [Google Scholar] [CrossRef]
- Tremellen, K.; Syedi, N.; Tan, S.; Pearce, K. Metabolic endotoxaemia–a potential novel link between ovarian inflammation and impaired progesterone production. Gynecol. Endocrinol. 2015, 4, 309–312. [Google Scholar] [CrossRef]
- Gnauck, A.; Lentle, R.G.; Kruger, M.C. Chasing a ghost?–Issues with the determination of circulating levels of endotoxin in human blood. Crit. Rev. Clin. Lab. Sci. 2016, 53, 197–215. [Google Scholar] [CrossRef]
- Grube, B.J.; Cochane, C.G.; Ye, R.D.; Green, C.E.; McPhail, M.E.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide binding protein expression in primary human hepatocytes and HepG2 hepatoma cells. J. Biol. Chem. 1994, 269, 8477–8482. [Google Scholar]
- Levin, J.; Bang., F. The role of endotoxin in the extracellular coagulation of Limulus blood. Bull. Johns Hopkins Hosp. 1964, 115, 265–274. [Google Scholar]
- Bergenson, A. Use of Lonza LAL Anaysis in Testing for Metabolic Endotoxemia; Global SME, Testing Solutions, Lonza: Walkersville, MD, USA, 2019. [Google Scholar]
- Clemente-Postigo, M.; Queipo-Ortuño, M.I.; Murri, M.; Boto-Ordoñez, M.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.J. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J. Lipid. Res. 2012, 53, 973–978. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential Effects of Cream, Glucose and Orange Juice on Inflammation, Endotoxin and the Expression of Toll Like Receptor-4 and Suppressor of Cytokine Signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Ghanim, H.; Sia, C.L.; Korzeniewski, K.; Lohano, T.; Abuaysheh, S.; Marumganti, A.; Chaudhuri, A.; Dandona, P. A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal. J. Clin. Endocrinol. Metab. 2011, 96, 1409–1414. [Google Scholar] [CrossRef]
- Ghanim, H.; Sia, C.L.; Upadhyay, M.; Korzeniewski, K.; Viswanathan, P.; Abuaysheh, S.; Mohanty, P.; Dandona, P. Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and Toll-like receptor expression. Am. J. Clin. Nutr. 2010, 91, 940–949. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; De Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef]
- Laugerette, F.; Vors, C.; Géloën, A.; Chauvin, M.-A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef]
- Roslansky, P.F.; Novitsky, T.J. Sensitivity of Limulus amebocyte lysate (LAL) to LAL-reactive glucans. J. Clin. Microbiol. 1991, 29, 2477–2483. [Google Scholar] [CrossRef]
- Cohen, J.; McConnell, J. Observations on the measurement and evaluation of endotoxemia by a quantitative limulus lysate microassay. J. Infect. Dis. 1984, 150, 916–924. [Google Scholar] [CrossRef]
- Pearson, F. Detection of endotoxin in the plasma of patients with gram-negative bacterial sepsis by the Limulus amoebocyte lysate assay. J. Clin. Micro. 1985, 21, 865–868. [Google Scholar] [CrossRef]
- Piya, M.K.; Harte, A.L.; McTernan, P.G. Metabolic endotoxaemia: Is it more than just a gut feeling? Curr. Opin. Lipidol. 2013, 24, 78–85. [Google Scholar] [CrossRef]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef]
- Laugerette, F.; Pineau, G.; Vors, C.; Michalski, M.C. Endotoxemia Analysis by the Limulus Amoebocyte Lysate Assay in Different Mammal Species Used in Metabolic Studies. J. Anal. Bioanal. Tech. 2015, 6, 4. [Google Scholar] [CrossRef]
- Gnauck, A.; Lentle, R.G.; Kruger, M.C. The Limulus Amebocyte Lysate assay may be unsuitable for detecting endotoxin in blood of healthy female subjects. J. Immunol. Methods 2015, 416, 146–156. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. BMI Classification. 2006. Available online: http://apps.who.int/bmi/index.jsp?introPage=intro_3.html. (accessed on 1 March 2020).
- Tremellen, K.; McPhee, N.; Pearce, K.; Benson, S.; Schedlowski, M.; Engler, H. Endotoxin-initiated inflammation reduces testosterone production in men of reproductive age. Am. J. Physiol. Endocrinol. Metabol. 2018, 314, E206–E213. [Google Scholar] [CrossRef] [PubMed]
- Tremellen, K.; Mcphee, N.; Pearce, K. Metabolic endotoxaemia related inflammation is associated with hypogonadism in overweight men. Basic Clin. Androl. 2017, 27, 5. [Google Scholar] [CrossRef] [PubMed]
- Meugnier, E.; Vidal, H.; Vors, C.; Laville, M.; Michalski, M.-C.; Pesenti, S.; Laugerette, F.; Pineau, G.; Drai, J.; Malpuech-Brugère, C. Postprandial Endotoxemia Linked With Chylomicrons and Lipopolysaccharides Handling in Obese Versus Lean Men: A Lipid Dose-Effect Trial. J. Clin. Endocrinol. Metabol. 2015, 100, 3427–3435. [Google Scholar] [CrossRef]
- Lopez-Moreno, J.; Garcia-Carpintero, S.; Gomez-Delgado, F.; Jimenez-Lucena, R.; Vals-Delgado, C.; Alcala-Diaz, J.F.; Roncero-Ramos, I.; Rangel-Zuñiga, O.A.; Yubero-Serrano, E.M.; Malagon, M.M.; et al. Endotoxemia is modulated by quantity and quality of dietary fat in older adults. Exp. Gerontol. 2018, 109, 119–125. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Ortega, F.; Serino, M.; Luche, E.; Waget, A.; Pardo, G.; Salvador, J.; Ricart, W.; Frühbeck, G.; Burcelin, R.; et al. Circulating lipopolysaccharide-binding protein (LBP) as a marker of obesity-related insulin resistance. Int. J. Obes. 2011, 36, 1442. [Google Scholar] [CrossRef]
- Bos, N.A.; Meeuwsen, C.G.; De Visser, H.; Benner, R. Clonal analysis of the synergistic mitogenic effect of lipopolysaccharide and dextran sulphate on B cell activation, growth, differentiation into Ig-secreting cells. Immunobiology 1988, 176, 301–312. [Google Scholar] [CrossRef]
- Czop, J.K.; Puglisi, A.V.; Miorandi, D.Z.; Austen, K.F. Perturbation of beta-glucan receptors on human neutrophils initiates phagocytosis and leukotriene B4 production. J. Immunol. 1988, 141, 3170–3176. [Google Scholar]
- Moore, J.N.; Cook, J.A.; Morris, D.D.; Halushka, P.V.; Wise, W.C. Endotoxin-induced procoagulant activity, eicosanoid synthesis, and tumor necrosis factor production by rat peritoneal macrophages: Effect of endotoxin tolerance and glucan. Circ. Shock. 1990, 31, 281–295. [Google Scholar]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef]
- Komuro, T.; Murai, T.; Kawasaki, H. Effect of sonication on the dispersion state of lipopolysaccharide and its pyrogenicity in rabbits. Chem. Pharm. Bull. 1987, 35, 4946–4952. [Google Scholar] [CrossRef]
- Yajima, Y.; Fukuda, I.; Otsuki, M.; Suzuki, H.; Ota, S.; Ishii, M.; Mori, K.; Goto, Y. Endotoxemia in liver diseases: Detection by a quantitative assay using chromogenic substrate with perchloric acid pretreatment. J. Exp. Med. 1985, 147, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Caroff, M.; Karibian, D. Structure of bacterial lipopolysaccharides. Carb. Res. 2003, 338, 2431–2447. [Google Scholar] [CrossRef] [PubMed]
- Pais de Barros, J.-P.; Gautier, T.; Sali, W.; Adrie, C.; Choubley, H.; Charron, E.; Lalande, C.; Le Guern, N.; Deckert, V.; Monchi, M.; et al. Quantitative lipopolysaccharide analysis using HPLC/MS/MS and its combination with the limulus amebocyte lysate assay. J. Lipid Res. 2015, 56, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Newhall, K.J.; Diemer, G.S.; Leshinsky, N.; Kerkof, K.; Chute, H.T.; Russell, C.B.; Rees, W.; Welcher, A.A.; Patterson, S.D.; Means, G.D. Evidence for Endotoxin Contamination in Plastic Na+-Heparin Blood Collection Tube Lots. Clin. Chem. 2010, 56, 1483–1491. [Google Scholar] [CrossRef]
- Sturk, A.; Joop, K.; ten Cate, J.W.; Thomas, L.L. Optimalization of a chromogenic assay for endotoxin in blood. Prog. Clin. Biol. Res. 1985, 189, 117–137. [Google Scholar]
- Roth, R.I.; Levin, F.C.; Levin, J. Optimization of detection of bacterial endotoxin in plasma with the Limulus test. J. Lab. Clin. Med. 1990, 116, 153–161. [Google Scholar]
- Levin, J.; Tomasulo, P.A.; Oser, R.S. Detection of endotoxin in human blood and demonstration of an inhibitor. J. Lab. Clin. Med. 1970, 75, 903–911. [Google Scholar]
- Harris, H.W.; Grunfeld, C.; Feingold, K.R.; Rapp, J.H. Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J. Clin. Investig. 1990, 86, 696–702. [Google Scholar] [CrossRef]
- Uddin, T.; Aktar, A.; Xu, P.; Johnson, R.A.; Rahman, M.A.; Leung, D.T.; Afrin, S.; Akter, A.; Alam, M.M.; Rahman, A.; et al. Immune Responses to O-Specific Polysaccharide and Lipopolysaccharide of Vibrio cholerae O1 Ogawa in Adult Bangladeshi Recipients of an Oral Killed Cholera Vaccine and Comparison to Responses in Patients with Cholera. Am. J. Trop. Med. Hygiene. 2014, 90, 873–881. [Google Scholar] [CrossRef][Green Version]
- Dullah, E.C.; Ongkudon, C.M. Current trends in endotoxin detection and analysis of endotoxin–protein interactions. Crit. Rev. Biotech. 2017, 37, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Ding, X. Methods of Endotoxin Detection. J. Lab. Automation. 2015, 20, 354–364. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample Pre-Treatment | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 70 °C/15 Mins | 70 °C/30 Mins | 80 °C/15 Mins | 80 °C/30 Mins | ||||||||||
| Sample | PPC | Sample | PPC | Sample | PPC | Sample | PPC | ||||||
| Dilution | CV | Rec | CV | CV | Rec | CV | CV | Rec | CV | CV | Rec | CV | |
| 1:50 | % | 100 | 40 | 100 | 100 | 33 | 100 | 100 | 33 | 100 | 100 | 40 | 100 |
| range | (1–8) | (28–62) | (1–6) | (1–4) | (35–188) | (1–5) | (1–4) | (37–67) | (1–15) | (1–6) | (37–67) | (1–8) | |
| 1:75 | % | 100 | 46 | 100 | 100 | 46 | 100 | 100 | 47 | 100 | 100 | 53 | 100 |
| range | (1–9) | (24–69) | (1–7) | (1–4) | (35–62) | (1–4) | (1–10) | (26–83) | (2–9) | (1–4) | (37–67) | (1–6) | |
| 1:100 | % | 100 | 53 | 100 | 100 | 46 | 100 | 100 | 53 | 100 | 100 | 60 | 100 |
| range | (1–6) | (38–96) | (1–10) | (1–23) | (22–90) | (1–3) | (1–10) | (24–103) | (2–6) | (1–14) | (35–93) | (1–8) | |
| 1:200 | % | 100 | 46 | 100 | 100 | 46 | 100 | 100 | 100 | 100 | 100 | 87 | 100 |
| range | (1–3) | (28–72) | (1–4) | (1–4) | (32–110) | (1–5) | (1–5) | (50–131) | (2–7) | (1–7) | (19–103) | (1–8) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearce, K.; Estanislao, D.; Fareed, S.; Tremellen, K. Metabolic Endotoxemia, Feeding Studies and the Use of the Limulus Amebocyte (LAL) Assay; Is It Fit for Purpose? Diagnostics 2020, 10, 428. https://doi.org/10.3390/diagnostics10060428
Pearce K, Estanislao D, Fareed S, Tremellen K. Metabolic Endotoxemia, Feeding Studies and the Use of the Limulus Amebocyte (LAL) Assay; Is It Fit for Purpose? Diagnostics. 2020; 10(6):428. https://doi.org/10.3390/diagnostics10060428
Chicago/Turabian StylePearce, Karma, Dianne Estanislao, Sinan Fareed, and Kelton Tremellen. 2020. "Metabolic Endotoxemia, Feeding Studies and the Use of the Limulus Amebocyte (LAL) Assay; Is It Fit for Purpose?" Diagnostics 10, no. 6: 428. https://doi.org/10.3390/diagnostics10060428
APA StylePearce, K., Estanislao, D., Fareed, S., & Tremellen, K. (2020). Metabolic Endotoxemia, Feeding Studies and the Use of the Limulus Amebocyte (LAL) Assay; Is It Fit for Purpose? Diagnostics, 10(6), 428. https://doi.org/10.3390/diagnostics10060428