Abstract
Endometriosis is characterized by ectopic endometrial tissue implantation, mostly within the peritoneum, and affects women in their reproductive age. Studies have been done to clarify its etiology, but the precise molecular mechanisms and pathophysiology remain unclear. We downloaded genome-wide mRNA expression and clinicopathological data of endometriosis patients and controls from NCBI’s Gene Expression Omnibus, after a systematic search of multiple independent studies comprising 156 endometriosis patients and 118 controls to identify causative genes, risk factors, and potential diagnostic/therapeutic biomarkers. Comprehensive gene expression meta-analysis, pathway analysis, and gene ontology analysis was done using a bioinformatics-based approach. We identified 1590 unique differentially expressed genes (129 upregulated and 1461 downregulated) mapped by IPA as biologically relevant. The top upregulated genes were FOS, EGR1, ZFP36, JUNB, APOD, CST1, GPX3, and PER1, and the top downregulated ones were DIO2, CPM, OLFM4, PALLD, BAG5, TOP2A, PKP4, CDC20B, and SNTN. The most perturbed canonical pathways were mitotic roles of Polo-like kinase, role of Checkpoint kinase proteins in cell cycle checkpoint control, and ATM signaling. Protein–protein interaction analysis showed a strong network association among FOS, EGR1, ZFP36, and JUNB. These findings provide a thorough understanding of the molecular mechanism of endometriosis, identified biomarkers, and represent a step towards the future development of novel diagnostic and therapeutic options.
1. Introduction
Endometriosis is a painful gynecological ailment marked by the presence of endometrial tissue outside the uterine cavity, commonly involving the uterus, ovaries, fallopian tubes, and pelvic tissues [1]. It is a complex and chronic estrogen-dependent disorder, wherein abnormal growth of uterine-lining (endometrium) tissue occurs outside the uterus, which can lead to serious complications like diabetes, obesity, mood disorders, dysmenorrhea, chronic pelvic pain, or even fatal endometrial cancer and cardiovascular disorders if left untreated for long. The most common site of endometriosis is the Douglas pouch (rectovaginal region) of the pelvic peritoneum [2]. Common symptoms include agonizing abdominal pain, period cramps (dysmenorrhea), heavy periods, pain with bowel movements or urination, dyspareunia, and infertility [3].
Pelvic exams and sonography are done to visualize abnormalities, and laparoscopy is required for diagnosis as well as treatment. Modes of treatment are primarily hormonal suppression and ultrasonographically guided surgical/laparoscopic management, which only provide symptomatic relief and the condition can recur with time [4]. Unfortunately, invasive surgery and the lack of a disease biomarker presently causes a mean latency of 7–11 years from symptom onset to definitive diagnosis. This substantial lag possibly has negative consequences in terms of disease progression.
An estimated 176 million women are affected by it worldwide, and studies suggest that 10% of females of reproductive age suffer from this inflammatory disorder. The prevalence of endometriosis is 11.1% among Saudi Arabian women [5]. However, the condition is often underdiagnosed and undertreated. Future higher risk for endometrial polyps [6] and rare progression to endometriosis-associated adenocarcinoma exists in endometriosis patients. They also have a lifetime predisposition to clear-cell and endometrioid types of ovarian cancer [7], which are endometriosis-derived, and which are possibly associated with retrograde menstruation [8]. The identification of a sufficiently specific and sensitive marker for the non-surgical detection of endometriosis would promise early diagnosis and prevention of detrimental effects, underscoring the need for comprehensive research. The most extensively studied potential biomarker for endometriosis is cancer antigen 125, but its use as a sole diagnostic marker is impractical due to its low sensitivity [9].
Recent innovations of high-throughput transcriptomics-based genome-wide approaches have had a major impact on medical research [10], thereby aiding in clinical classification and treatment predictions [10,11,12]. Understanding the genetic basis of the pathophysiology of endometriosis is important to explain the strong genetic association with heritability, estimated at around 50% [13]. Dysregulation of several genes has been implicated in the etiology of this ectopic condition.
In a menstrual cycle, endometrium undergoes transition from estrogen-dominant proliferative (follicular) phases (early-proliferative (EP), mid-proliferative (MP), and late-proliferative (LP)) to progesterone-dominant secretory (luteal) phases ((early-secretory (ES), mid-secretory (MS), and late-secretory (LS)), followed by the menstrual phase. A distinct differential transcriptional profile exists for each endometrial cycle phase [14,15]. As the uterine linings in endometriosis patients have altered transcriptomic profiles, molecular classification is needed for disease identification and staging [16]. Previous works have focused on expression profiling of different stages of the endometrial cycle in small groups. Herein, we integrated data to conduct comprehensive differential transcriptional profiling of a large cohort in order to identify differentially expressed genes, upstream regulators, and perturbed canonical pathways that could possibly be used in future to identify novel potential biomarkers and therapeutic targets for endometriosis.
Etiology
Decrease in age of menarche, fewer pregnancies, less breast feeding, and increase in maternal age at first birth all cause an overall increase in the number of ovulations and menstruations within a reproductive lifespan. These changes are associated strongly with the risk of endometriosis development and tend to be more pronounced during the decade of highest risk for endometriosis, i.e., 25–35 years of age [17]. Estrogen dependence, immune modulation, and certain environmental pollutants, mostly dioxins and polychlorinated biphenyls, perhaps contribute to the disease’s pathobiology [18]. Immunological or hormonal dysfunction make some women predisposed to endometriosis. Higher macrophage activation and humoral immune responsiveness with reduced cell-mediated immunity, with weakened T-cell and NK-cell responses, are seen in women suffering from endometriosis. Humoral autoantibodies against endometrial and ovarian tissue have been detected in endometriosis patient sera [19].
The pathogenesis of endometriosis has been speculated to result from aberrant angiogenesis that occurs in the eutopic endometrium with retrograde menstruation—“Sampson’s hypothesis” [2]. Factors that increase the rate of retrograde menstruation, such as congenital outflow tract obstructions, might also predispose to endometriosis. Detailed understanding on the basis of gene expression studies is lacking, and findings are often inconsistent or even contradictory.
2. Materials and Methods
2.1. Data Retrieval and Sample Description
Our approach was the integration of publicly available gene expression data generated by different microarray platforms. We first retrieved whole-transcript array datasets (.CEL files) along with provided clinical details of endometriosis patients dated up to 30 March 2020 from the Gene Expression Omnibus (GEO, NCBI) databank, a public domain hosting high-throughput genomic data. The present study included following expression data series with GEO accession numbers GSE7846, GSE7305, GSE6364, GSE4888, GSE51981, GSE31683, and GSE25628 and their sample information to compare the transcriptomic status of affected and control patients (Table 1). The GSE51981 dataset has a total of 148 endometrial samples from patients with ages ranging from 20–50 years. It includes samples from women in different menstrual cycle phases, including endometriosis with severe pelvic pain/infertility (n = 77) and normal without endometriosis (n = 71). Normal women with uterine fibroids, adenomyosis, or pelvic organ prolapse were further grouped as normal with uterine/pelvic pathology (n = 37), and others as normal without uterine pathology (n = 34). The GSE7846 dataset includes five arrays for human endometrial endothelial cells (HEECs) derived from eutopic endometria of patients with endometriosis, and five from patients without endometriosis (controls). GSE7305 includes expression profiles of 10 each of normal and diseased cases.
2.2. Gene Expression Analysis
To generate expression profiles of endometriosis samples, .CEL files were imported to Partek Genomics Suite, version 7.0 (Partek Inc., St. Louis, MO, USA) followed by log-transformation and normalization of the robust background-adjusted array dataset. Principal component analysis (PCA) was done on high-dimensional data to assess quality and overall variance in gene expression of individuals among sample groups. Analysis of variance (ANOVA) was employed to create a list of differentially expressed genes (DEGs) with a cut-off p-value of ≤ 0.05 and fold change of ± 2. Hierarchical clustering was done to reveal the pattern of most differentially expressed (up- and downregulated) genes across samples.
2.3. Gene Ontology, Pathway, and Upstream Regulators Analysis
The identified statistically significant DEGs with corresponding probe sets ID, p-value, fold-change values, and other relevant data were uploaded into the Ingenuity Pathways Analysis (IPA, QIAGEN’s Ingenuity Systems, Redwood City, CA, USA) software for molecular network and canonical pathway analysis to define interaction amongst the differentially regulated genes using functional algorithms. The Benjamini–Hochberg method was used to adjust p-values for canonical pathways, and p-values below 0.01 and Altman z-scores of ± 2 were considered significant. Positive and negative values of z-score represent activation and inhibition of dysregulated canonical pathways. Gene ontology study was done to functionally categorize endometriosis-significant genes. All endometriosis-associated DEGs were imported to figuratively represent all identified connections and potential relationships among them, in order to identify significant pathways leading to endometriosis initiation and progression.
2.4. Protein–Protein Interaction Analysis
To check the interactions at the protein level, the STRING v11.0 database (http://string-db.org) was used to search for possible physical and functional associations among proteins encoded by the top DEGs (including both up- and downregulated) for a better understanding of disease pathobiology [20]. This prediction gives a visual idea about the possible interconnections between the proteins involved in a specific disease network.
3. Results
Differentially Expressed Genes from Meta-Analysis
Integration of the seven GEO data series included in present study comprised a total of 156 endometriosis patients and 118 controls. Data were merged before analysis as all had used the same GPL570 platform, except GSE25628 which used the GPL571 platform (Table 1).

Table 1.
Gene expression Omnibus (GEO) data series, platform, and sample description of endometrium-based expression studies (total 156 endometriosis patients + 118 normal cases).
Principal component analysis showed the grouping of the samples in three-dimensional space as per their whole-genome expression patterns, where each circle represents an individual (Figure 1). Comparing endometriosis with normal non-endometriosis tissue without any pelvic/uterine pathology resulted in the detection of 1590 differentially expressed genes (129 upregulated and 1461 downregulated). The top upregulated genes, including FOS, EGR1, ZFP36, JUNB, APOD, CST1, GPX3, and PER1, are shown in Table 2 and the top downregulated genes, including DIO2, CPM, OLFM4, PALLD, BAG5, TOP2A, PKP4, CDC20B, and SNTN, are shown in Table 3. Hierarchical clustering of DEGs showed a clear difference in expression pattern of genes between endometriosis cases and controls (Figure 2). Disease and functional annotation of DEGs broadly predicted endometrial adenocarcinoma. However, DEGs like FOS, EGR1, ZFP36, JUNB, GPX3, PAEP, DUSP1, MT1M, COL6A1, NR4A1, TGFB1, CITED2, IL2RG, ACKR1, JUN, PTGER3, COL6A2, PGR, PLK2, PLA2G4A, FBN1, MPPED2, EZR, MMP11, GALNT4, PTEN, PIK3CA, CREB1, ERBIN, DNMT3A, REL, SDC2, ZNF25, ITGA6, GUCY1A2, PDGFD, OVGP1, ITGB1, APOBEC3B, OLFM1, NRIP1, MEF2A, CNTN1, BUB1B, MEST, KIF20A, RRM1, ANK3, and CCNA2 showed significant association with endometriosis (p-value = 0.0006).
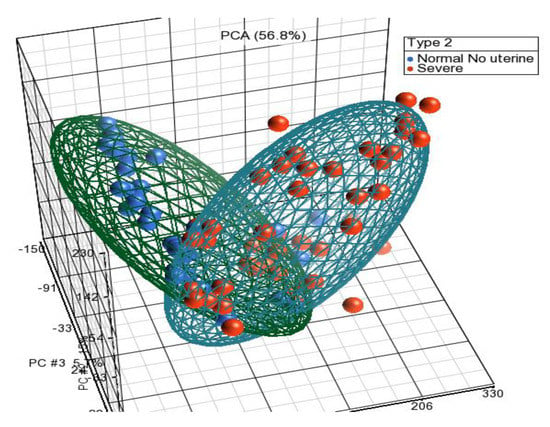
Figure 1.
PCA showing two clear distinct clusters for severe endometriosis cases and normal healthy controls without uterine pathology.
Table 2.
Top overexpressed/upregulated differentially expressed genes in endometriosis.
Table 3.
The top downregulated differentially expressed genes in meta-analysis of endometriosis.
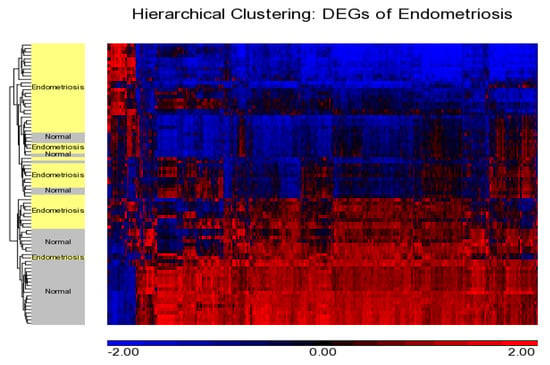
Figure 2.
Hierarchical clustering showing distribution of DEGs and cases. Downregulated and upregulated genes are shown in blue and red, respectively, showing a distinct pattern with a majority of genes found to be downregulated in endometriosis.
Ingenuity pathway analysis for the DEGs of endometriosis revealed altered canonical pathways that were either activated or inhibited (Figure 3, Table 4). Mitotic roles of polo-like kinase (z-score −2.71), aldosterone signaling in epithelial cells (z-score −3.464), and role of CHK proteins in cell cycle checkpoint control (z-score −0.632) were found to be inhibited while ATM signaling (z-score + 1.698) and SUMOylation pathways (z-score + 2.668) were activated (Figure 4, Figure 5). IPA predicted the activation status of upstream regulators among identified DEGs of endometriosis. REL (transcription factor, z-score −4.13, Pval 0.0002), CTNNB1 (transcription factor, z-score −3.2, Pval 0.01), PGR (ligand-dependent nuclear receptor, z-score −2.2, Pval 0.0005), and VCAN (proteoglycan, z-score −2.6, Pval 0.02) were the top inhibited upstream regulators (Table 5). We also used a biological database, STRING, to predict functional associations and interaction between the proteins encoded by the identified significant DEGs (top up- and downregulated ones), and the results are shown in Figure 6. The network indicated a strong interplay of various proteins and their specific involvement in endometriosis.
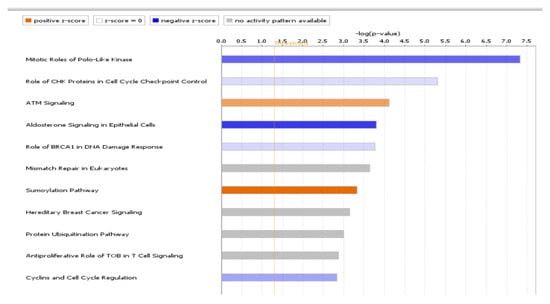
Figure 3.
Significant pathways identified by IPA. The top 11 altered canonical pathways predicted from DEGs of endometriosis. Negative and positive z-scores, colored in shades of blue and red, represent inhibition and activation of pathways, respectively.
Table 4.
Significant canonical pathways based on DEGs of endometriosis. Positive and negative z- scores indicate overall activation and inhibition status of pathways, respectively.
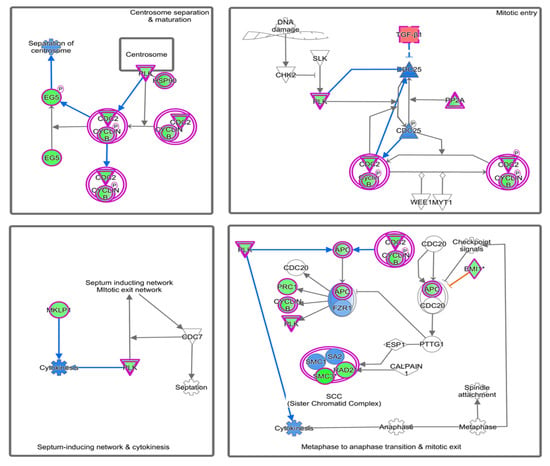
Figure 4.
Significant inhibition of mitotic polo-like kinase pathway predicted by IPA. Pathway genes were overlaid to the DEGs; upregulated (TGFB1, CDC25, FZR1) genes are shown in red/pink, while downregulated genes (PRC1, PLK, Cyclin B1, SMC3) are shown in green.
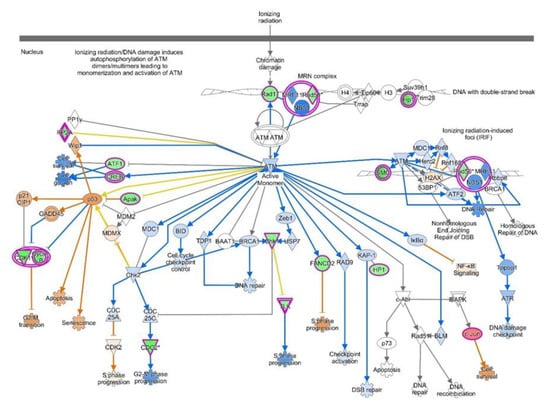
Figure 5.
Activation of ATM signaling predicted by IPA-based pathway analysis. Pathway genes were overlaid to the DEGs, with upregulated ones shown in shades of red and downregulated ones shown in green.
Table 5.
Most significant upstream regulators and target molecules in endometriosis dataset.
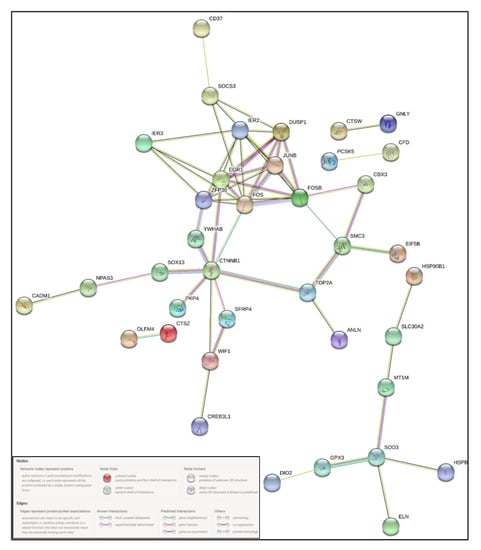
Figure 6.
STRING-based protein–protein interaction (PPI) network of selected differentially expressed genes, showing predicted interconnectivities.
4. Discussion
Endometriosis, a growth/deposition of endometrial tissue at extra-uterine sites, affects around 10% of reproductive women. In addition to abnormal reproductive physiological problems, cases are increasing drastically due to adverse consequences of treatment with oral contraceptives, GnRH agonists, synthetic progestins, and aromatase inhibitors (letrozole) to prevent the menstrual cycle and/or pregnancy [1,21]. Understanding the molecular etiology of origin and progression of endometriosis is necessary to explore therapeutic options and provide better treatment. We therefore conducted transcriptomic meta-analysis to identify endometriosis-associated significant DEGs and essential pathological pathways.
Combining multiple studies has always been challenging, as different studies use varied protocols, platforms, and analysis methods. We used raw data (.CEL) files to integrate multiple data series to get a bigger cohort and analyzed the data. We identified transcriptomic signatures of endometriosis and evaluated the roles of specific genes, upstream regulators, and dysregulated pathways. Our results provide some insight into the molecular mechanisms underlying endometriosis pathogenesis. Pathogenic genes and pathways may serve as novel targets for diagnostic and prognostic biomarkers and potential therapies for endometriosis. In the present study, we had a long list of genes and pathways, but have restricted our discussion to the most prominent genes and pathways.
4.1. Molecular Etiology of Endometriosis
Retrograde or “reverse” menstruation has been suggested as an initial cause of endometriosis, where menstrual blood is thrown back into pelvic cavity outside the uterus, instead of flowing out of the cervix. This endometrial tissue growth out of the uterus is the result of an estrogen-dependent hormonal local imbalance. Higher prevalence has been also seen in women with immune disorders (like rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, and hypo- or hyperthyroidism) [17]. Recently, a small-molecule agonist G-1 (Tespria) against the G-protein-coupled estrogen receptor also showed reduction in endometrial growth [22].
Unusual transformation of certain abdominal wall cells into endometrial cells has been reported in some women [23] and, interestingly, it is believed that during embryonic development, the same cells are responsible for the growth of female reproductive organs. Researchers also think that pelvic inflammation, damage, or infection of cells that line the pelvis like a prior caesarean surgery can also trigger endometriosis [23,24,25]. The exact pathogenesis still remains uncertain. We therefore conducted a transcriptomics study in order to understand the genetic factors that allow cells to grow as endometrial tissue outside the uterus.
In our results, we found high expression of early and immediate early-response genes such as FBJ murine osteosarcoma viral oncogene homolog or Fos proto-Oncogene (FOS), FosB Proto-Oncogene (FOSB), Early Growth Response 1 (EGR1), ZFP36 Ring Finger Protein (ZFP36), Immediate Early Response 2 (IER2), Immediate Early Response 3 (IER3), Jun B Proto-Oncogene (JUNB), and Transcription Factor SOX-13 (SOX 13). The majority of these are DNA-binding proteins that act as transcriptional factors. Some others, like Dual specificity protein phosphatase 1 (DUSP1) and Receptor-type tyrosine-protein phosphatase O (PTPRO), possess phosphatase activity.
c-Fos is the transcription factor of the Fos family, including FosB, Fra-1, and Fra-2 [26]. It is an immediate early-response gene involved in cell proliferation and differentiation of normal tissue after extracellular stress stimuli. Its deregulation has been linked to oncogenic transformation and tumor progression. FOS plays a significant role in endometrial cells’ proliferation and its overexpression is associated with a poor prognosis of endometrial carcinoma [27]. Fos and Jun family proteins form a heterodimer complex of AP-1 transcription factor, shown to be involved in endometrial carcinogenesis [28]. Upstream regulator analysis revealed genes such as REL, CTNNB1, PGR, and VCAN by analyzing linkage to DEGs that were experimentally shown to affect gene expression [29]. All upstream regulators were inhibited.
REL:REL (V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog) encodes for the proto-oncogene c-Rel protein, a transcription factor of the NF-κB family that regulates genes involved in B- and T-cell differentiation, immune response, survival, apoptosis, proliferation, and oncogenic processes, including endometrial carcinogenesis [30,31].
CTNNB1:CTNNB1 (Catenin β1) codes for a protein that regulates and coordinates cell–cell adhesion, embryonic development, epithelial–mesenchymal transition, and gene transcription. It is an integral part of the canonical Wnt pathway. Aberrant Wnt/β-catenin signaling pathway function is allied with loose cytoskeleton organization and cell-to-cell contacts of epithelial cells, along with a high motility of mesenchymal cells to promote invasiveness and fibrosis. This might lead to multiple cancers, including endometrial cancer [32,33,34,35,36]. Targeting the Wnt/β-catenin signaling was shown to avert fibrogenesis in a xenograft endometriosis mice model [35].
VCAN:VCAN (Versican) codes for four extracellular matrix isoforms like large chondroitin sulfate proteoglycan in different tissues and organs that regulate cell adhesion, proliferation, migration, and survival [2]. Higher expression of VCAN has been reported in angiogenesis, tumor growth, cancer relapse, and inflammatory lung disorders [37,38,39]. Significantly high expression of VCAN was also reported in the mid-secretory phase of endometrial epithelial cells after combination estrogen/progesterone treatment. The V1 isoform of VCAN was recently reported to the facilitate development of endometrial receptivity and human embryo implantation [40]. Higher expression of VCAN is connected with pathogenesis of peritoneal endometriosis and seems to be an indicator of poor prognosis endometrial cancer [2,41].
Alteration in expression of HOXB4 [42], apelin peptide [43], interleukin 18 [44], estrogen and progesterone receptors [45], integrin β3 and osteopontin (OPN) [46], microRNA-29c, and FKBP4 [47] have been reported. Varied expression levels of metastasis-inducing proteins (S100P, S100A4, OPN, and anterior gradient homologue 2 (AGR2)) have been shown to enhance pathogenesis by increasing endometrial cell invasiveness and establishing endometriotic ectopic deposits after retrograde menstruation [48].
Aromatase activates estrogen biosynthesis locally from androgens, thereby sequentially stimulating a positive feedback cycle of prostaglandin E2 production by upregulating cyclooxygenase-2 (COX-2). Because of lack of aromatase (estrogen synthase) in the normal endometrium, androgens cannot be converted into estrogen [49]. In contrast, numerous studies have described aberrantly high expression of aromatase in eutopic and ectopic endometrium [17]. Increased COX-2 expression in the stromal cells and aberrant aromatase overexpression in eutopic endometrium have both been indicated as potential therapeutic biomarkers, and therefore, their specific inhibitors are being increasingly employed for therapeutic management [50]. A probable connection of Krüppel-like Factor 9 (KLF9) dysregulation has been suggested in both pregnancy failure and endometrial pathogenesis [51]. The progesterone resistance and subsequent infertility seen in endometriosis seems to have an association with KLF9, a progesterone-receptor-interacting protein, as mice null for Klf9 are sub-fertile. It is implicated that deficiency of KLF9 contributes to progesterone resistance of eutopic endometrium in patients [52] and exhibits simultaneous abrogation of Hedgehog-, Notch-, and steroid-receptor-regulated networks [53].
Based on serum proteomic differential expression, a possible biomarker panel comprising zinc-alpha-2-glycoprotein, albumin, and complement C3 has been proposed for effective and non-invasive diagnosis of endometriosis [54]. Importantly, the three markers were independent from the endometriosis stage and cycle phase. Brain Derived Neurotrophic Factor (BDNF) has been identified as a potential peripheral early diagnostic marker, as its mean plasma concentrations were twice as high in endometriosis cases in contrast to asymptomatic or healthy controls [9]. Based on this, a nano-chip-based electrochemical detection technique was developed. The only limitation to this is its non-specificity, as the variations in BDNF expression have been reported in numerous unconnected pathologies [55].
4.2. Canonical Pathways Involved in Endometriosis
Molecular pathway analysis revealed a couple of significantly altered canonical pathways for DEGs of endometriosis. Herein, we discuss the role of key pathways like Mitotic roles of polo-like kinase, Role of CHK proteins in cell cycle checkpoint control, Aldosterone signaling in epithelial cells, and ATM Signaling in endometriosis progression.
Mitotic Roles of Polo-Like Kinase Pathway: The Polo-like kinase (Plks) is a member of the serine/threonine protein kinase (PLK1-5) family that regulates the mitotic checkpoint during M phase of cell division. Plks can act either as oncogene or tumor suppressor, and has been found to be overexpressed in different cancer types including endometrial [56] and ovarian [57] cancers. Because of its direct association with increased cellular proliferation and poor prognosis, it is considered a bona fide cancer biomarker [58,59]. Direct association of Plks expression with serum estrogen (ovarian hormone) levels and abnormal regulation of ectopic endometrial cell proliferation strongly suggest its role in the pathogenesis of endometriosis [60]. Plks inhibitors such as volasertib and rigosertib are in advanced stage of clinical trials and might be used for endometriosis treatment [61].
Role of CHK Proteins in Cell Cycle Checkpoint Control Pathway: Activation of cell cycle checkpoint kinases including Chk1 and Chk2 are an instant response to repair any type of DNA damage [62]. In response to DNA damage, this signaling pathway temporarily delays cell cycle progression, allowing time for DNA repair, or triggers programmed cell death. Activated ATM kinase phosphorylates Chk2 which phosphorylates CDC25C to block the progression from G2 to M phase. Chk2 also phosphorylates p53, attenuating p53 binding to MDM2 and activating p21/WAF1 to arrest the G1 phase of the cell cycle. Rad3-dependent activation of Chk1 phosphorylates CDC25A and CDC2 to inhibit their activity to block G2–M transition. Overall, CHK protein signaling depends on the type of stress and extent of DNA damage and is involved in endometrial cancer [63].
Cisplatin exerts an anticancer effect by activating DNA-damage-response genes Chk1/2, which generates both survival (repair) and apoptotic signals that lead to cell death. Cisplatin-resistant cells have dominant repair signaling that allows cells to survive. Chk1/2 inhibitor AZD7762 has been shown to overcome cisplatin resistance in endometriosis-associated ovarian cancer by reducing repair signaling [64].
ATM Signaling Pathway: Ataxia telangiectasia mutated (ATM) gene codes for serine/threonine protein kinase and participates in cell division and DNA repair. DNA damage induces autophosphorylation of ATM which activates DNA repair enzymes by phosphorylating Chk1/2 to fix the broken strands [65]. Efficient cross-talk between ATR-Chk1 and ATM-Chk2 leads to repair of damaged DNA strands which helps to maintain the cell’s genomic stability and integrity [66]. The ATM signaling pathway, because of its central role in cell division and DNA repair, has been a focus of cancer research, especially endometrial cancer, for exploring novel molecular therapies targeting ATM pathways [67].
Aldosterone Signaling in Epithelial Cells Pathway: Aldosterone is a mineralocorticoid steroid hormone produced by the adrenal cortex. Aldosterone signaling primarily controls blood pressure and inflammation by regulating its target genes (FKBP5, IGF1, KRAS, PKCε, NCOA1, NCOR1, NEDD4L, SGK, and MR/NR3C2 as per RGD, https://rgd.mcw.edu [68] and IPA). Recent studies have shown the possible involvement of aldosterone in multiple gynecological problems and inflammatory disorders [69]. There is a well-established association of endometriosis with intraperitoneal inflammation diseases like atherosclerosis and hypertension, and also with autoimmune diseases like diabetes, hypothyroidism, and cancer [9]. A metabolomics-based study revealed high aldosterone levels in endometriosis patients with infertility [70].
4.3. Future Directions
The strength of present work lies in the inclusion of multiple endometriosis-related expression datasets in order to understand endometriosis at the molecular level. However, the absence of a validation study was its limitation. In future, we plan to conduct RT-PCR-based validation studies for differentially expressed genes on endometriosis samples collected from the Jeddah region. Further cell cultures and animal models could be used to assess the effect of activated/suppressed genes on molecular pathways and disease phenotypes for potential clinical translation. Virtual screening of potential lead compounds against identified therapeutic biomarkers for rational drug design will be done. This could facilitate imminent tailor-made personalized therapies.
5. Conclusions
Endometriosis is an estrogen-dependent, progesterone-resistant, inflammatory multifactorial gynecological disorder. Identification of distinct molecular signatures and potential therapeutic molecules corresponding to endometriosis is needed for better diagnosis. The present microarray-based genomics and molecular pathway analysis method helped to establish a better understanding of endometriosis at the molecular level, as multiple expression datasets were integrated to determine differentially expressed genes and identify canonical molecular pathways related to endometriosis in a broad way. The study identified alterations of gene expression and molecular signaling, including aldosterone signaling, that result in the hormonal imbalances and pathogenesis of endometriosis. An anti-inflammatory diet and increased levels of antioxidants and phytonutrients can be recommended to patients to reverse inflammation and oxidative damage, while also supporting healthy hormone balance.
Author Contributions
Z.M. and U.A.A.-D. Study design, U.A.A.-D. Acquisition of data, Z.M. Analysis and interpretation of data, Z.M. Manuscript writing, U.A.A.-D. Critical review of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This Project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no. 31217 (Code: G: 500-141-1439). The authors, therefore, acknowledge and thanks DSR for its technical and financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cho, Y.J.; Lee, S.H.; Park, J.W.; Han, M.; Park, M.J.; Han, S.J. Dysfunctional signaling underlying endometriosis: Current state of knowledge. J. Mol. Endocrinol. 2018, 60, R97–R113. [Google Scholar] [CrossRef]
- Tani, H.; Sato, Y.; Ueda, M.; Miyazaki, Y.; Suginami, K.; Horie, A.; Konishi, I.; Shinomura, T. Role of Versican in the Pathogenesis of Peritoneal Endometriosis. J. Clin. Endocrinol. Metab. 2016, 101, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Alimi, Y.; Iwanaga, J.; Loukas, M.; Tubbs, R.S. The Clinical Anatomy of Endometriosis: A Review. Cureus 2018, 10, e3361. [Google Scholar] [CrossRef] [PubMed]
- Yeung, P., Jr. The laparoscopic management of endometriosis in patients with pelvic pain. Obstet. Gynecol. Clin. N. Am. 2014, 41, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Rouzi, A.A.; Sahly, N.; Kafy, S.; Sawan, D.; Abduljabbar, H. Prevalence of endometriosis at a university hospital in Jeddah, Saudi Arabia. Clin. Exp. Obstet. Gynecol. 2015, 42, 785–786. [Google Scholar]
- Zheng, Q.M.; Mao, H.I.; Zhao, Y.J.; Zhao, J.; Wei, X.; Liu, P.S. Risk of endometrial polyps in women with endometriosis: A meta-analysis. Reprod. Biol. Endocrinol. 2015, 13, 103. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, T.H.; Chung, H.H.; Song, Y.S. Risk and prognosis of ovarian cancer in women with endometriosis: A meta-analysis. Br. J. Cancer 2014, 110, 1878–1890. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Hughes, C.; Foster, W.; Agarwal, S. The impact of endometriosis across the lifespan of women: Foreseeable research and therapeutic prospects. Biomed. Res. Int. 2015, 2015, 158490. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Hedenfalk, I.; Duggan, D.; Chen, Y.; Radmacher, M.; Bittner, M.; Simon, R.; Meltzer, P.; Gusterson, B.; Esteller, M.; Kallioniemi, O.P.; et al. Gene-expression profiles in hereditary breast cancer. N. Engl. J. Med. 2001, 344, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Elledge, R.; Mohsin, S.; Osborne, C.K.; Chamness, G.C.; Allred, D.C.; et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 2003, 362, 362–369. [Google Scholar] [CrossRef]
- Borghese, B.; Zondervan, K.T.; Abrao, M.S.; Chapron, C.; Vaiman, D. Recent insights on the genetics and epigenetics of endometriosis. Clin. Genet. 2017, 91, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ponnampalam, A.P.; Weston, G.C.; Trajstman, A.C.; Susil, B.; Rogers, P.A.W. Molecular classification of human endometrial cycle stages by transcriptional profiling. Mol. Hum. Reprod. 2004, 10, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, R.; Catalano, R.; Sharkey, A. Large-scale gene expression studies of the endometrium: What have we learnt? Reproduction 2006, 132, 1–10. [Google Scholar] [CrossRef][Green Version]
- Miller, L.M.; Johnson, N.P. EPHect—The Endometriosis Phenome (and Biobanking) Harmonisation Project—May be very helpful for clinicians and the women they are treating. F1000Res 2017, 6, 14. [Google Scholar] [CrossRef]
- Parazzini, F.; Vercellini, P.; Pelucchi, C. Endometriosis: Epidemiology, and Etiological Factors. In Endometriosis; Linda, C.G., Johannes, L.H.E., David, L.H., Eds.; Wiley-Blackwell: Oxford, UK, 2012; pp. 19–26. [Google Scholar] [CrossRef]
- Porpora, M.G.; Resta, S.; Fuggetta, E.; Storelli, P.; Megiorni, F.; Manganaro, L.; De Felip, E. Role of environmental organochlorinated pollutants in the development of endometriosis. Clin. Exp. Obstet. Gynecol. 2013, 40, 565–567. [Google Scholar]
- Mathur, S.; Peress, M.R.; Williamson, H.O.; Youmans, C.D.; Maney, S.A.; Garvin, A.J.; Rust, P.F.; Fudenberg, H.H. Autoimmunity to endometrium and ovary in endometriosis. Clin. Exp. Immunol. 1982, 50, 259–266. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Attar, E.; Bulun, S.E. Aromatase inhibitors: The next generation of therapeutics for endometriosis? Fertil. Steril. 2006, 85, 1307–1318. [Google Scholar] [CrossRef]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Silva, T.S.; Dias, M.F. Clear cell carcinoma arising from abdominal wall endometriosis - Brief report and review of the literature. Gynecol. Oncol. Rep. 2017, 20, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım, D.; Tatar, C.; Doğan, O.; Hut, A.; Dönmez, T.; Akıncı, M.; Toptaş, M.; Bayık, R.N. Post-cesarean scar endometriosis. Turk. J. Obstet. Gynecol. 2018, 15, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Durán, E.E.; Aboharp-Hasan, Z.; Mendoza-Morales, R.C.; García-Rodríguez, F.M.; Jiménez-Villanueva, X.; Peñavera-Hernández, J.R. Clear cell adenocarcinoma arising from abdominal wall endometriosis. Cirugía Cir. 2016, 84, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Nemos, C.; Delage-Mourroux, R.; Jouvenot, M.; Adami, P. Onset of direct 17-beta estradiol effects on proliferation and c-fos expression during oncogenesis of endometrial glandular epithelial cells. Exp. Cell Res. 2004, 296, 109–122. [Google Scholar] [CrossRef]
- Mahner, S.; Baasch, C.; Schwarz, J.; Hein, S.; Wölber, L.; Jänicke, F.; Milde-Langosch, K. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. Br. J. Cancer 2008, 99, 1269–1275. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef]
- Pallares, J.; Martínez-Guitarte, J.L.; Dolcet, X.; Llobet, D.; Rue, M.; Palacios, J.; Prat, J.; Matias-Guiu, X. Abnormalities in the NF-κB family and related proteins in endometrial carcinoma. J. Pathol. 2004, 204, 569–577. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. beta-catenin signaling and cancer. Bioessays News Rev. Mol. Cell. Dev. Biol. 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Chen, Y.; Song, W. Wnt/catenin β1/microRNA 183 predicts recurrence and prognosis of patients with colorectal cancer. Oncol Lett. 2018, 15, 4451–4456. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Darcha, C. Involvement of the Wnt/beta-Catenin Signaling Pathway in the Cellular and Molecular Mechanisms of Fibrosis in Endometriosis. PLoS ONE 2013, 8, e76808. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Yang, W.-X. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017, 8, 41679–41689. [Google Scholar] [CrossRef]
- Kischel, P.; Waltregny, D.; Dumont, B.; Turtoi, A.; Greffe, Y.; Kirsch, S.; De Pauw, E.; Castronovo, V. Versican overexpression in human breast cancer lesions: Known and new isoforms for stromal tumor targeting. Int. J. Cancer 2010, 126, 640–650. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Yee, A.J. Roles of versican in cancer biology—Tumorigenesis, progression and metastasis. Histol. Histopathol. 2013, 28, 701–713. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; Hallgren, O.; Rolandsson, S.; Weitoft, M.; Tykesson, E.; Larsson-Callerfelt, A.K.; Rydell-Törmänen, K.; Bjermer, L.; Malmström, A.; Karlsson, J.C.; et al. Versican in inflammation and tissue remodeling: The impact on lung disorders. Glycobiology 2015, 25, 243–251. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Horie, A.; Tani, H.; Ueda, M.; Okunomiya, A.; Suginami, K.; Kondoh, E.; Baba, T.; Konishi, I.; Shinomura, T.; et al. Versican V1 in human endometrial epithelial cells promotes BeWo spheroid adhesion in vitro. Reproduction 2019, 157, 53. [Google Scholar] [CrossRef]
- Kodama, J.; Kusumoto, T.; Seki, N.; Matsuo, T.; Ojima, Y.; Nakamura, K.; Hongo, A.; Hiramatsu, Y. Prognostic significance of stromal versican expression in human endometrial cancer. Ann. Oncol. 2007, 18, 269–274. [Google Scholar] [CrossRef]
- AlKusayer, G.M.; Pon, J.R.; Peng, B.; Klausen, C.; Lisonkova, S.; Kinloch, M.; Yong, P.; Muhammad, E.M.S.; Leung, P.C.K.; Bedaiwy, M.A. HOXB4 Immunoreactivity in Endometrial Tissues From Women With or Without Endometriosis. Reprod. Sci. 2017. [Google Scholar] [CrossRef]
- Ozkan, Z.S.; Cilgin, H.; Simsek, M.; Cobanoglu, B.; Ilhan, N. Investigation of apelin expression in endometriosis. J. Reprod. Infertil. 2013, 14, 50–55. [Google Scholar]
- Luo, Q.; Ning, W.; Wu, Y.; Zhu, X.; Jin, F.; Sheng, J.; Huang, H. Altered expression of interleukin-18 in the ectopic and eutopic endometrium of women with endometriosis. J. Reprod. Immunol. 2006, 72, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.K.; Bulmer, J.N.; Searle, R.F. Immunohistochemical characterization of proliferation, oestrogen receptor and progesterone receptor expression in endometriosis: Comparison of eutopic and ectopic endometrium with normal cycling endometrium. Hum. Reprod. 1995, 10, 3272–3279. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yang, X.L.; Sun, X.; Peng, C.; Li, X.; Wang, M.; Zhou, Y.F. [Expression of integrin beta3 and osteopontin in endometrium of patients with adenomyosis]. Zhonghua Fu Chan Ke Za Zhi 2009, 44, 354–358. [Google Scholar] [PubMed]
- Joshi, N.R.; Miyadahira, E.H.; Afshar, Y.; Jeong, J.W.; Young, S.L.; Lessey, B.A.; Serafini, P.C.; Fazleabas, A.T. Progesterone Resistance in Endometriosis Is Modulated by the Altered Expression of MicroRNA-29c and FKBP4. J. Clin. Endocrinol. Metab. 2017, 102, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hapangama, D.K.; Raju, R.S.; Valentijn, A.J.; Barraclough, D.; Hart, A.; Turner, M.A.; Platt-Higgins, A.; Barraclough, R.; Rudland, P.S. Aberrant expression of metastasis-inducing proteins in ectopic and matched eutopic endometrium of women with endometriosis: Implications for the pathogenesis of endometriosis. Hum. Reprod. 2012, 27, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Yang, S.; Fang, Z.; Gurates, B.; Tamura, M.; Zhou, J.; Sebastian, S. Role of aromatase in endometrial disease. J. Steroid Biochem. Mol. Biol. 2001, 79, 19–25. [Google Scholar] [CrossRef]
- Ebert, A.D.; Bartley, J.; David, M. Aromatase inhibitors and cyclooxygenase-2 (COX-2) inhibitors in endometriosis: New questions—Old answers? Eur. J. Obstet. Gynecol. Reprod. Biol. 2005, 122, 144–150. [Google Scholar] [CrossRef]
- Simmen, F.A.; Su, Y.; Xiao, R.; Zeng, Z.; Simmen, R.C.M. The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod. Biol. Endocrinol. 2008, 6, 41. [Google Scholar] [CrossRef]
- Pabona, J.M.; Simmen, F.A.; Nikiforov, M.A.; Zhuang, D.; Shankar, K.; Velarde, M.C.; Zelenko, Z.; Giudice, L.C.; Simmen, R.C. Kruppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: Implications for the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E376–E392. [Google Scholar] [CrossRef] [PubMed]
- Heard, M.E.; Simmons, C.D.; Simmen, F.A.; Simmen, R.C. Kruppel-like factor 9 deficiency in uterine endometrial cells promotes ectopic lesion establishment associated with activated notch and hedgehog signaling in a mouse model of endometriosis. Endocrinology 2014, 155, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Signorile, P.G.; Baldi, A. Looking for an effective and non-invasive diagnostic test for endometriosis: Where are we? Ann. Transl. Med. 2018, 17, S106. [Google Scholar] [CrossRef] [PubMed]
- Bockaj, M.; Fung, B.; Tsoulis, M.; Foster, W.G.; Soleymani, L. Method for Electrochemical Detection of Brain Derived Neurotrophic Factor (BDNF) in Plasma. Anal. Chem. 2018, 90, 8561–8566. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Polo-like kinase (PLK) expression in endometrial carcinoma. Cancer Lett. 2001, 169, 41–49. [Google Scholar] [CrossRef]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett. 2001, 164, 41–49. [Google Scholar] [CrossRef]
- de Cárcer, G. The Mitotic Cancer Target Polo-Like Kinase 1: Oncogene or Tumor Suppressor? Genes 2019, 10, 208. [Google Scholar] [CrossRef]
- Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers (Basel) 2017, 9, 131. [Google Scholar] [CrossRef]
- Tang, L.; Wang, T.-T.; Wu, Y.-T.; Zhou, C.-Y.; Huang, H.-F. High expression levels of cyclin B1 and Polo-like kinase 1 in ectopic endometrial cells associated with abnormal cell cycle regulation of endometriosis. Fertil. Steril. 2009, 91, 979–987. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef]
- Zhou, B.-B.S.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, V.; Millar, A.; Briollais, L.; Chapman, W.; Bapat, B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002, 62, 4095–4099. [Google Scholar] [PubMed]
- Itamochi, H.; Nishimura, M.; Oumi, N.; Kato, M.; Oishi, T.; Shimada, M.; Sato, S.; Naniwa, J.; Sato, S.; Kudoh, A.; et al. Checkpoint Kinase Inhibitor AZD7762 Overcomes Cisplatin Resistance in Clear Cell Carcinoma of the Ovary. Int. J. Gynecol. Cancer 2014, 24, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, J.-H.; Paull, T.T.; Feigenbaum, L.; Nussenzweig, A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J. Cell. Biol. 2008, 183, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Mun Tho, L.; Xu, N.; A. Gillespie, D. Chapter 3—The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 108, pp. 73–112. [Google Scholar]
- Takeuchi, M.; Tanikawa, M.; Nagasaka, K.; Oda, K.; Kawata, Y.; Oki, S.; Agapiti, C.; Sone, K.; Miyagawa, Y.; Hiraike, H.; et al. Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers (Basel) 2019, 11, 1913. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Hayman, G.T.; Wang, S.J.; Laulederkind, S.J.F.; Hoffman, M.J.; Kaldunski, M.L.; Tutaj, M.; Thota, J.; Nalabolu, H.S.; Ellanki, S.L.R.; et al. The Year of the Rat: The Rat Genome Database at 20: A multi-species knowledgebase and analysis platform. Nucleic Acids Res. 2020, 48, D731–D742. [Google Scholar] [CrossRef]
- Sabbadin, C.; Andrisani, A.; Ambrosini, G.; Bordin, L.; Donà, G.; Manso, J.; Ceccato, F.; Scaroni, C.; Armanini, D. Aldosterone in Gynecology and Its Involvement on the Risk of Hypertension in Pregnancy. Front. Endocrinol. 2019, 10, 575. [Google Scholar] [CrossRef]
- Letsiou, S.; Peterse, D.P.; Fassbender, A.; Hendriks, M.M.; van den Broek, N.J.; Berger, R.; Dorien, F.O.; Vanhie, A.; Vodolazkaia, A.; Van Langendonckt, A.; et al. Endometriosis is associated with aberrant metabolite profiles in plasma. Fertil. Steril. 2017, 107, 699–706.e696. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).