Novel Splice Site Pathogenic Variant of EFTUD2 Is Associated with Mandibulofacial Dysostosis with Microcephaly and Extracranial Symptoms in Korea



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical and Audiologic Evaluation
2.3. Chromosomal and Targeted Gene Studies
2.4. Exome Sequencing, Variant Calling, and Variant Annotation
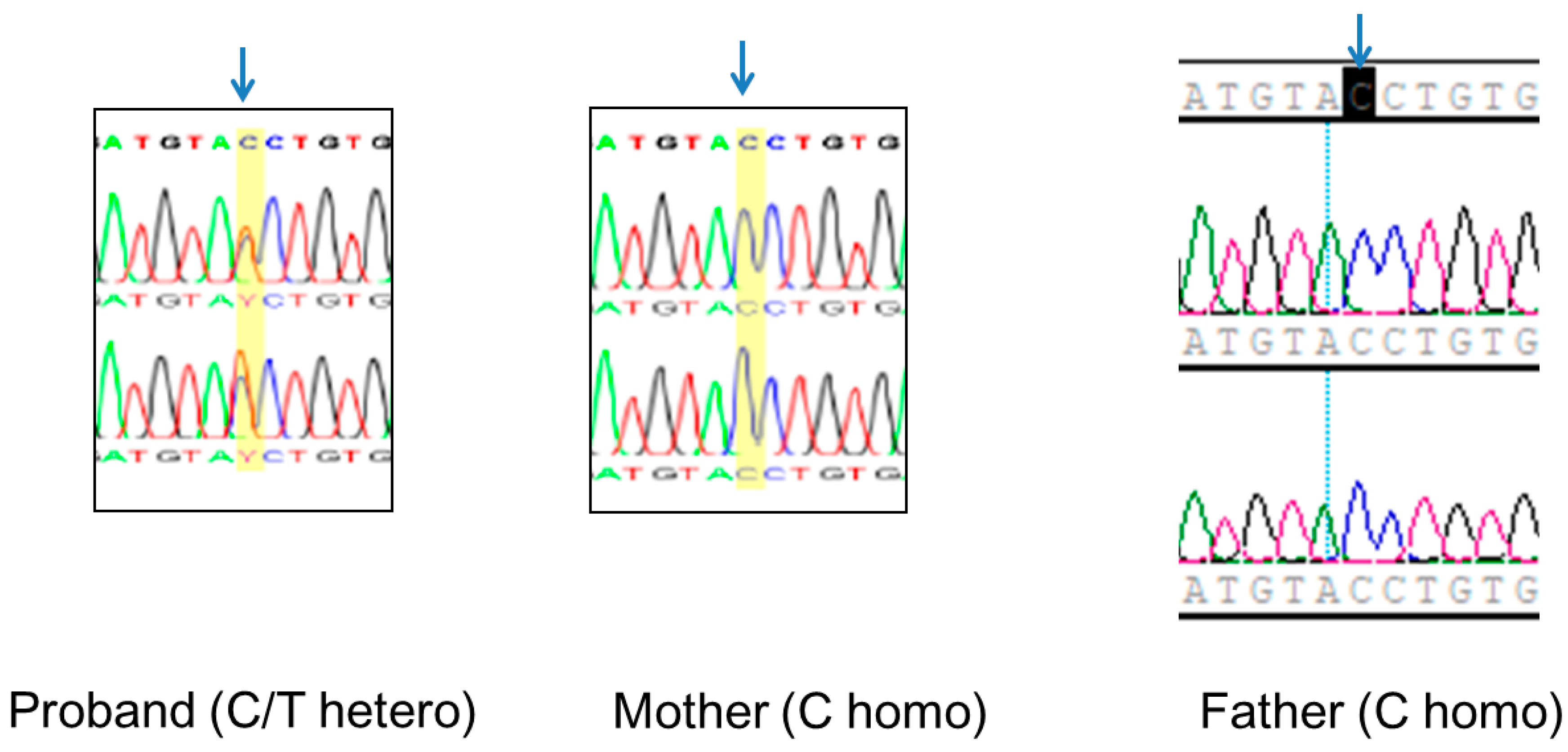
2.5. Validation of the EFTUD2 Variant and TRIO Studies
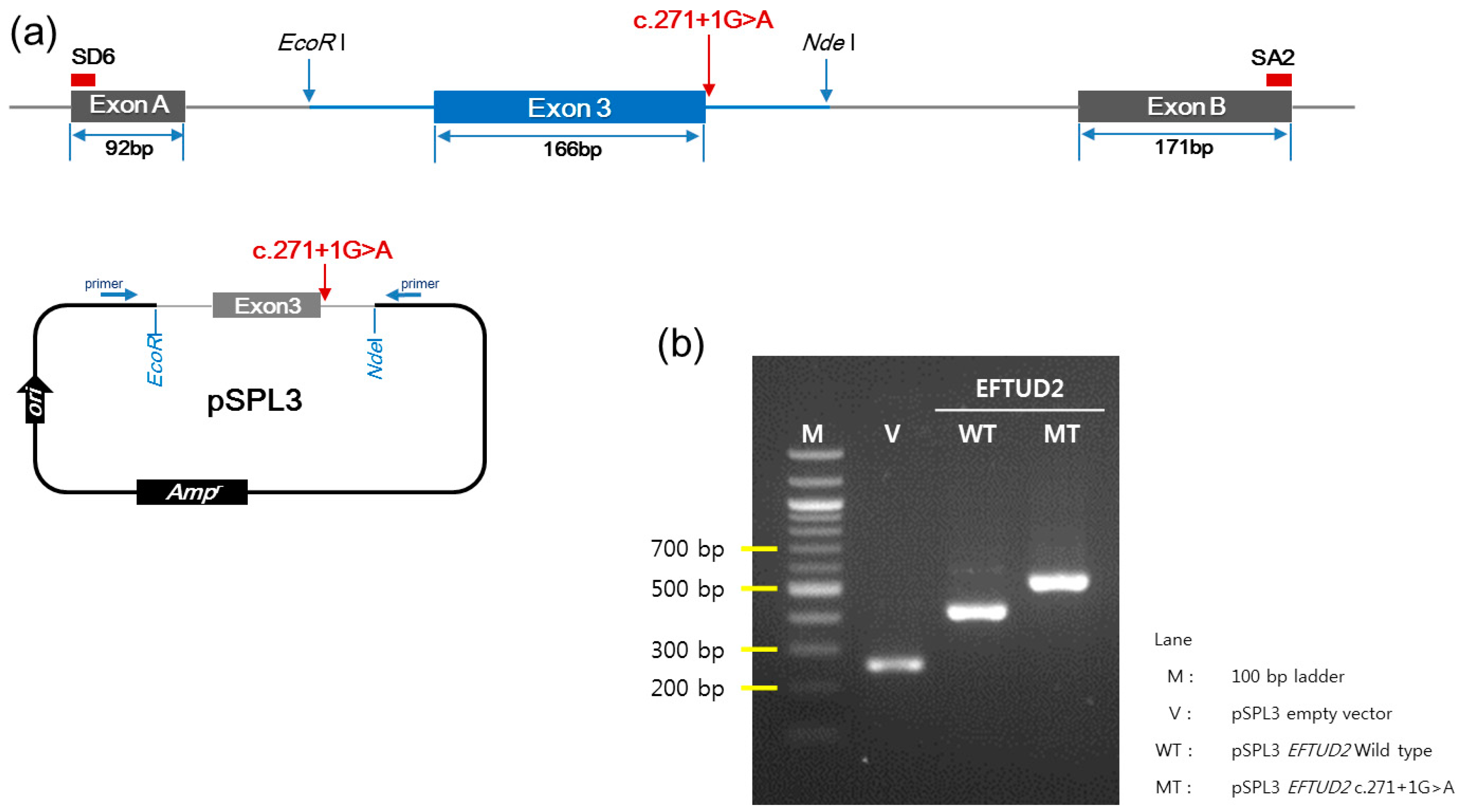
2.6. Minigene Assay
3. Results
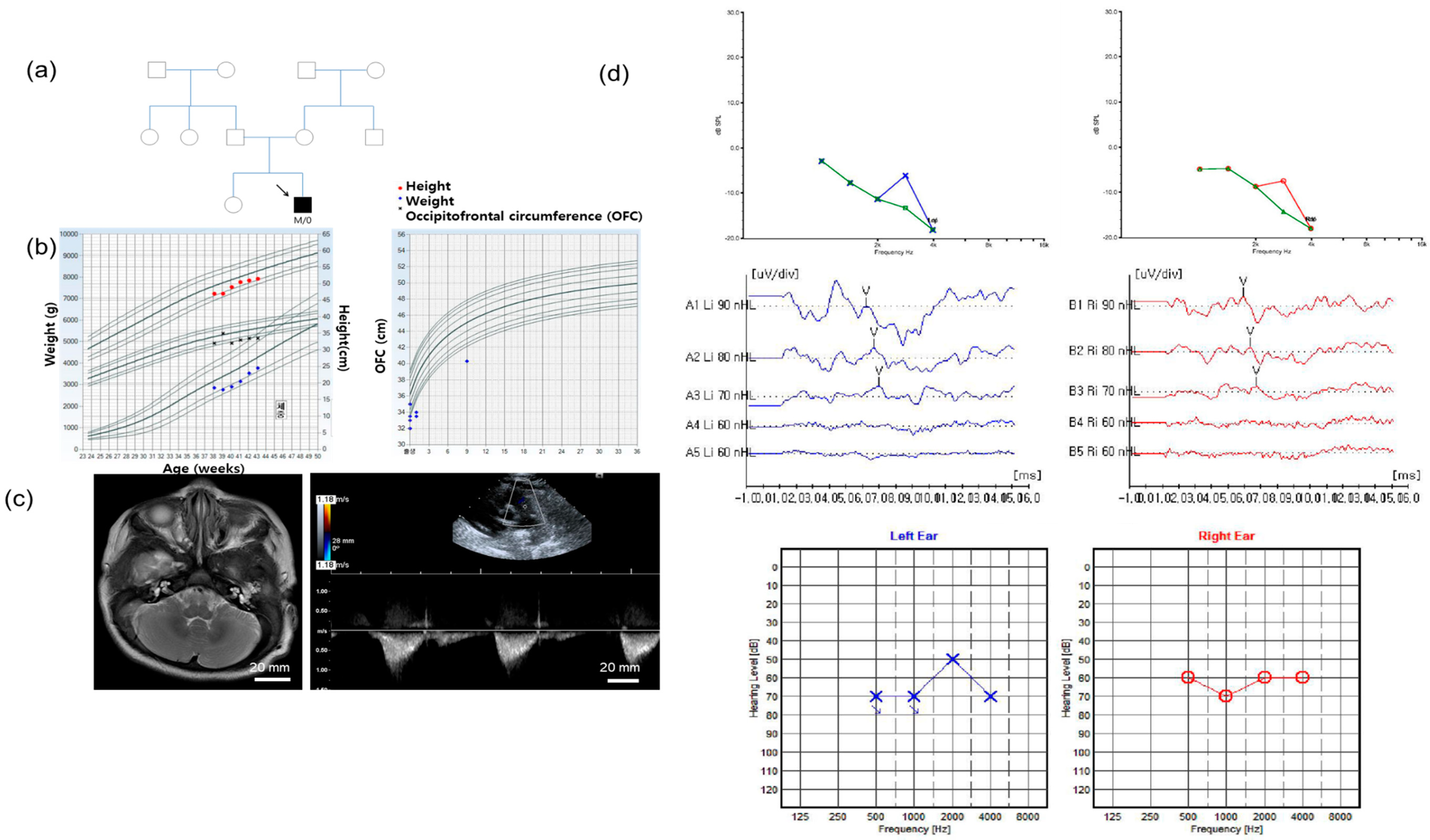
3.1. Clinical Manifestations
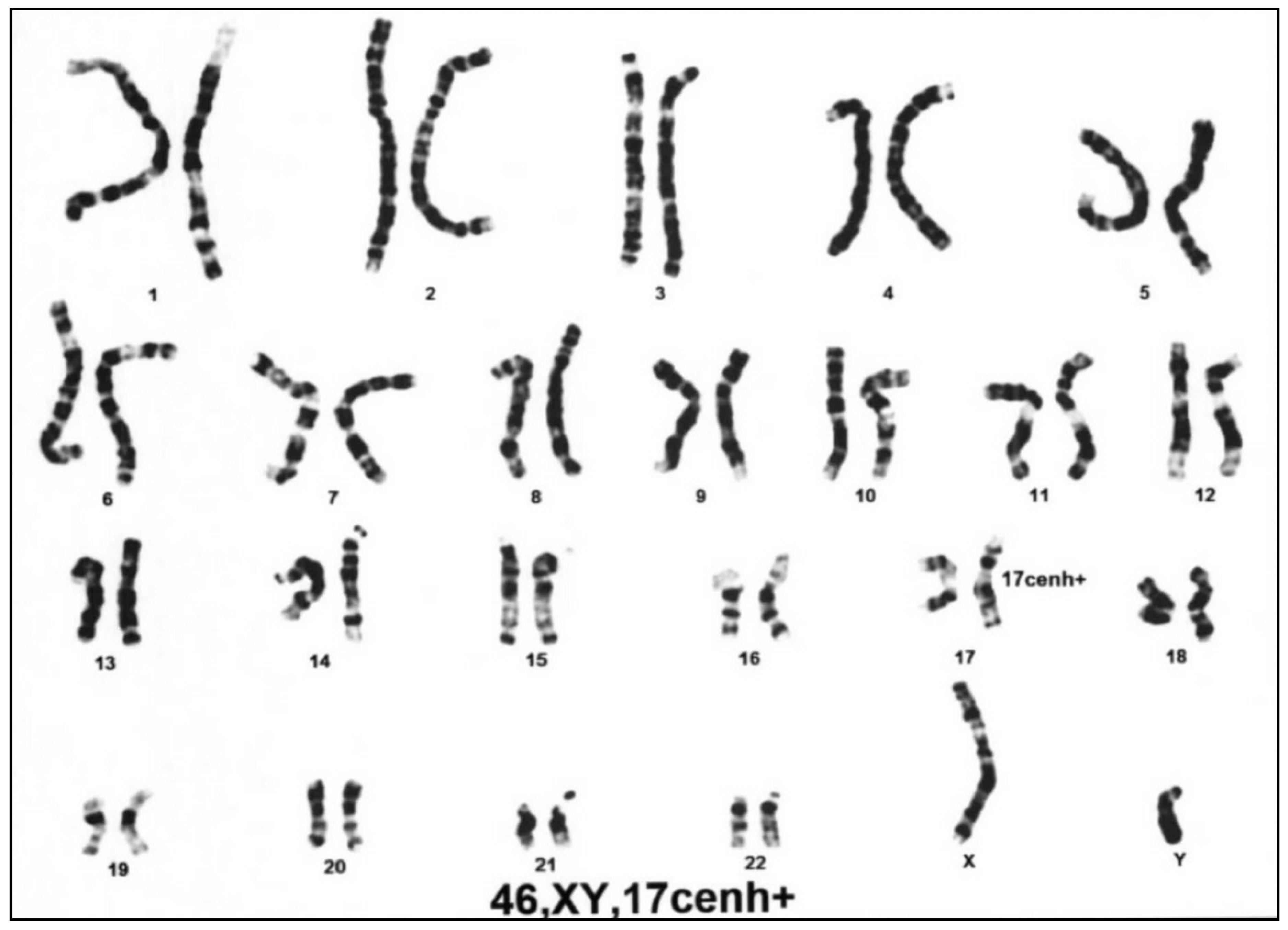
3.2. Differential Molecular Genetic Diagnosis
3.3. Identification of a Novel Splice Site EFTUD2 Variant
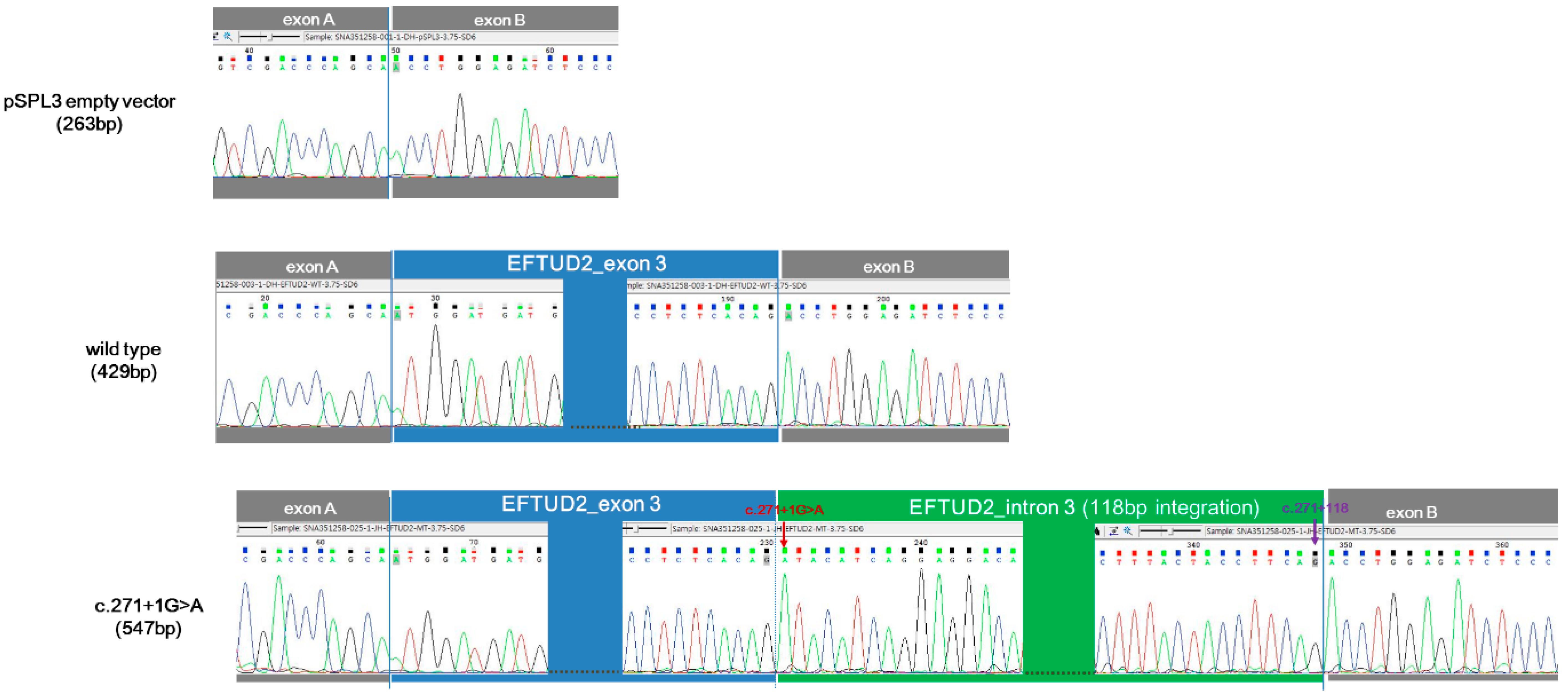
3.4. In Vitro Functional Characterization of c.271+1G>A
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guion-Almeida, M.L.; Zechi-Ceide, R.M.; Vendramini, S.; Tabith Junior, A. A new syndrome with growth and mental retardation, mandibulofacial dysostosis, microcephaly, and cleft palate. Clin. Dysmorphol. 2006, 15, 171–174. [Google Scholar] [CrossRef]
- Lines, M.A.; Huang, L.; Schwartzentruber, J.; Douglas, S.L.; Lynch, D.C.; Beaulieu, C.; LeineGuion-Almeida, M.; MariaZechi-Ceide, R.; Gener, B.; Gillessen-Kaesbach, G.; et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 2012, 90, 369–377. [Google Scholar] [CrossRef]
- Huang, L.; Vanstone, M.R.; Hartley, T.; Osmond, M.; Barrowman, N.; Allanson, J.; Baker, L.; Dabir, T.A.; Dipple, K.M.; Dobyns, W.B.; et al. Mandibulofacial Dysostosis with Microcephaly: Mutation and Database Update. Hum. Mutat. 2016, 37, 148–154. [Google Scholar] [CrossRef]
- Silva, J.B.; Soares, D.; Leao, M.; Santos, H. Mandibulofacial dysostosis with microcephaly: A syndrome to remember. BMJ Case Rep. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Luquetti, D.V.; Hing, A.V.; Rieder, M.J.; Nickerson, D.A.; Turner, E.H.; Smith, J.; Park, S.; Cunningham, M.L. “Mandibulofacial dysostosis with microcephaly” caused by EFTUD2 mutations: Expanding the phenotype. Am. J. Med. Genet. Part A 2013, 161A, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Emrick, L.T.; Smith, E.M.; Austin, E.G.; Yang, Y.; Hunter, J.V.; Scaglia, F.; Lalani, S.R. Novel de novo mutations in EFTUD2 detected by exome sequencing in mandibulofacial dysostosis with Microcephaly syndrome. Am. J. Med. Genet. Part A 2015, 167A, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Yamauchi, A.; Ito, Y.; Sakauchi, M.; Yamamoto, T.; Okamoto, N.; Tsurusaki, Y.; Miyake, N.; Matsumoto, N.; Saito, K. Mandibulofacial dysostosis with microcephaly: A case presenting with seizures. Brain Dev. 2017, 39, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Bezniakow, N.; Jakubiak, A.; Bloch, M.; Patkowski, D.; Obersztyn, E.; Sasiadek, M.M. Phenotype analysis of Polish patients with mandibulofacial dysostosis type Guion-Almeida associated with esophageal atresia and choanal atresia caused by EFTUD2 gene mutations. J. Appl. Genet. 2015, 56, 199–204. [Google Scholar] [CrossRef]
- Wieczorek, D.; Gener, B.; Gonzalez, M.J.; Seland, S.; Fischer, S.; Hehr, U.; Hoefsloot, L.H.; de Leeuw, N.; Gillessen-Kaesbach, G. Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay in three unrelated patients: A mandibulofacial dysostosis distinct from Treacher Collins syndrome. Am. J. Med. Genet. Part A 2009, 149A, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef]
- Rengasamy Venugopalan, S.; Farrow, E.G.; Lypka, M. Whole-exome sequencing identified a variant in EFTUD2 gene in establishing a genetic diagnosis. Orthod. Craniofac. Res. 2017, 20 (Suppl. 1), 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lacour, J.C.; McBride, L.; St Hilaire, H.; Mundinger, G.S.; Moses, M.; Koon, J.; Torres, J.I.; Lacassie, Y. Novel De Novo EFTUD2 Mutations in 2 Cases with MFDM, Initially Suspected to Have Alternative Craniofacial Diagnoses. Cleft Palate Craniofac. J. 2019, 56, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; Nguyen, P.D.; Oh, D.Y.; Han, J.H.; Kim, A.R.; Kim, M.Y.; Park, H.-R.; Tran, L.H.; Dung, N.H.; Koo, J.-W.; et al. Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population. Sci. Rep. 2019, 9, 1604. [Google Scholar] [CrossRef]
- Lee, S.Y.; Joo, K.; Oh, J.; Han, J.H.; Park, H.R.; Lee, S.; Oh, D.-Y.; Woo, S.J.; Choi, B.Y. Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation. Clin. Exp. Otorhinolaryngol. 2019. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Voigt, C.; Megarbane, A.; Neveling, K.; Czeschik, J.C.; Albrecht, B.; Callewaert, B.; von Deimling, F.; Hehr, A.; Smeland, M.F.; König, R.; et al. Oto-facial syndrome and esophageal atresia, intellectual disability and zygomatic anomalies—Expanding the phenotypes associated with EFTUD2 mutations. Orphanet J. Rare Dis. 2013, 8, 110. [Google Scholar] [CrossRef]
- Lehalle, D.; Gordon, C.T.; Oufadem, M.; Goudefroye, G.; Boutaud, L.; Alessandri, J.L.; Baena, N.; Baujat, G.; Baumann, C.; Boute-Benejean, O.; et al. Delineation of EFTUD2 haploinsufficiency-related phenotypes through a series of 36 patients. Hum. Mutat. 2014, 35, 478–485. [Google Scholar] [CrossRef]
- Yu, K.P.T.; Luk, H.M.; Gordon, C.T.; Fung, G.; Oufadem, M.; Garcia-Barcelo, M.M.; Amiel, J.; Chung, B.H.Y.; Lo, I.F.M.; Tiong, Y.T. Mandibulofacial dysostosis Guion-Almeida type caused by novel EFTUD2 splice site variants in two Asian children. Clin. Dysmorphol. 2018, 27, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Bick, D.; Fraser, P.C.; Gutzeit, M.F.; Harris, J.M.; Hambuch, T.M.; Helbling, D.C.; Jacob, H.J.; Kersten, J.N.; Leuthner, S.R.; May, T.; et al. Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J. Pediatr. Genet. 2017, 6, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.T.; Petit, F.; Oufadem, M.; Decaestecker, C.; Jourdain, A.S.; Andrieux, J.; Malan, V.; Alessandri, J.; Baujat, G.; Baumann, C.; et al. EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J. Med. Genet. 2012, 49, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.S.; Ingles, J.; Semsarian, C.; Bagnall, R.D. Key Value of RNA Analysis of MYBPC3 Splice-Site Variants in Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002368. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef]
- Booth, K.T.; Askew, J.W.; Talebizadeh, Z.; Huygen, P.L.M.; Eudy, J.; Kenyon, J.; Hoover, D.; Hildebrand, M.S.; Smith, K.R.; Bahlo, M.; et al. Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37. Genet. Med. 2019, 21, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Dufner-Almeida, L.G.; do Carmo, R.T.; Masotti, C.; Haddad, L.A. Understanding human DNA variants affecting pre-mRNA splicing in the NGS era. Adv. Genet. 2019, 103, 39–90. [Google Scholar] [CrossRef]
- Small, E.C.; Leggett, S.R.; Winans, A.A.; Staley, J.P. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol. Cell 2006, 23, 389–399. [Google Scholar] [CrossRef]
- Bartels, C.; Klatt, C.; Luhrmann, R.; Fabrizio, P. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep. 2002, 3, 875–880. [Google Scholar] [CrossRef]
- Lei, L.; Yan, S.Y.; Yang, R.; Chen, J.Y.; Li, Y.; Bu, Y.; Chang, N.; Zhou, Q.; Zhu, X.; Li, C.Y.; et al. Spliceosomal protein eftud2 mutation leads to p53-dependent apoptosis in zebrafish neural progenitors. Nucleic Acids Res. 2017, 45, 3422–3436. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Y.; He, Y.; Li, Q.; Wang, X.; Sun, C.; Wang, L.; An, Y.; Luo, F. EFTUD2 gene deficiency disrupts osteoblast maturation and inhibits chondrocyte differentiation via activation of the p53 signaling pathway. Hum. Genom. 2019, 13, 63. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.Y.; Lee, D.-h.; Han, J.H.; Choi, B.Y. Novel Splice Site Pathogenic Variant of EFTUD2 Is Associated with Mandibulofacial Dysostosis with Microcephaly and Extracranial Symptoms in Korea. Diagnostics 2020, 10, 296. https://doi.org/10.3390/diagnostics10050296
Kim SY, Lee D-h, Han JH, Choi BY. Novel Splice Site Pathogenic Variant of EFTUD2 Is Associated with Mandibulofacial Dysostosis with Microcephaly and Extracranial Symptoms in Korea. Diagnostics. 2020; 10(5):296. https://doi.org/10.3390/diagnostics10050296
Chicago/Turabian StyleKim, So Young, Da-hye Lee, Jin Hee Han, and Byung Yoon Choi. 2020. "Novel Splice Site Pathogenic Variant of EFTUD2 Is Associated with Mandibulofacial Dysostosis with Microcephaly and Extracranial Symptoms in Korea" Diagnostics 10, no. 5: 296. https://doi.org/10.3390/diagnostics10050296
APA StyleKim, S. Y., Lee, D.-h., Han, J. H., & Choi, B. Y. (2020). Novel Splice Site Pathogenic Variant of EFTUD2 Is Associated with Mandibulofacial Dysostosis with Microcephaly and Extracranial Symptoms in Korea. Diagnostics, 10(5), 296. https://doi.org/10.3390/diagnostics10050296