Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features


{kind=link}
{kind=link}
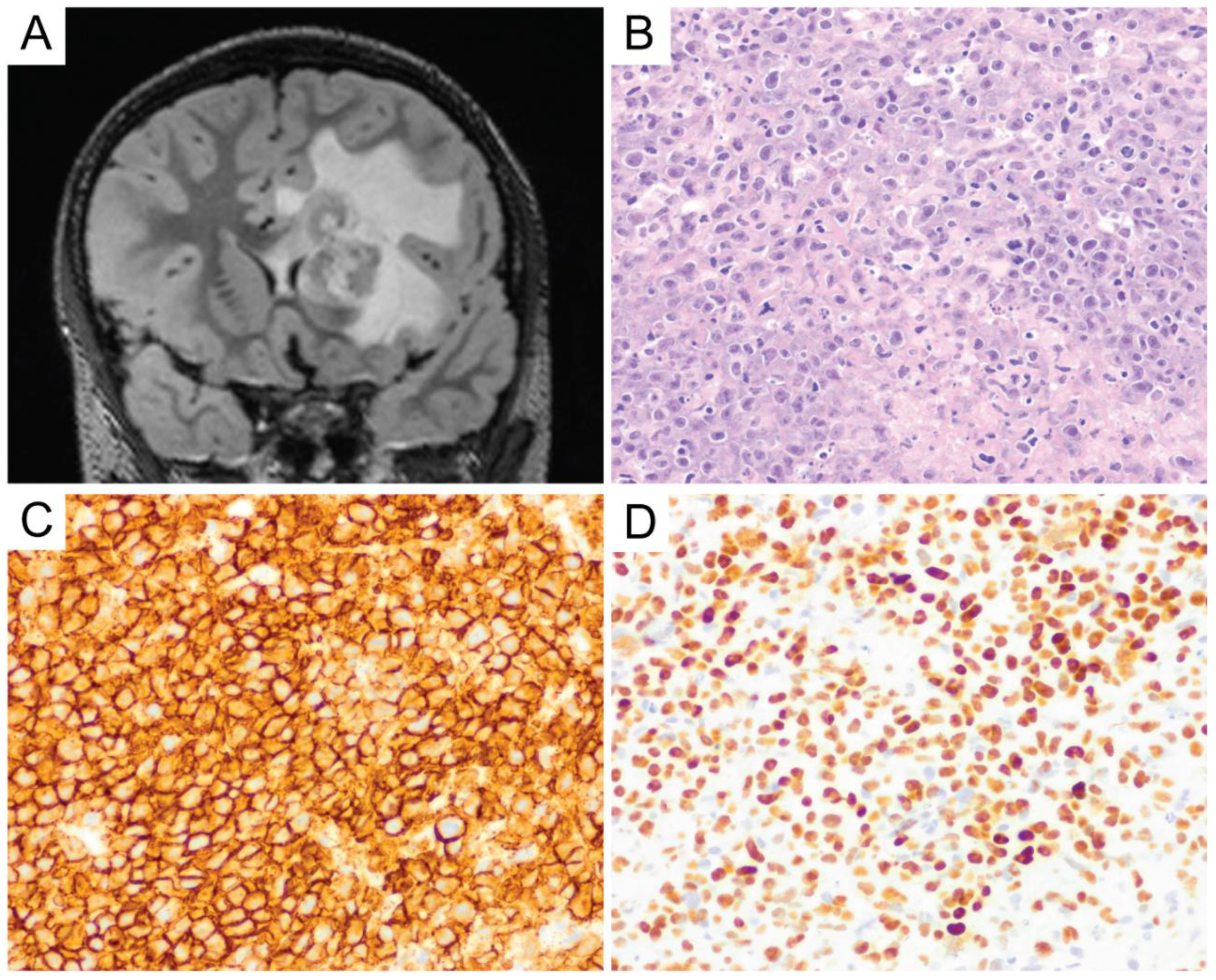{kind=link}
{kind=link}
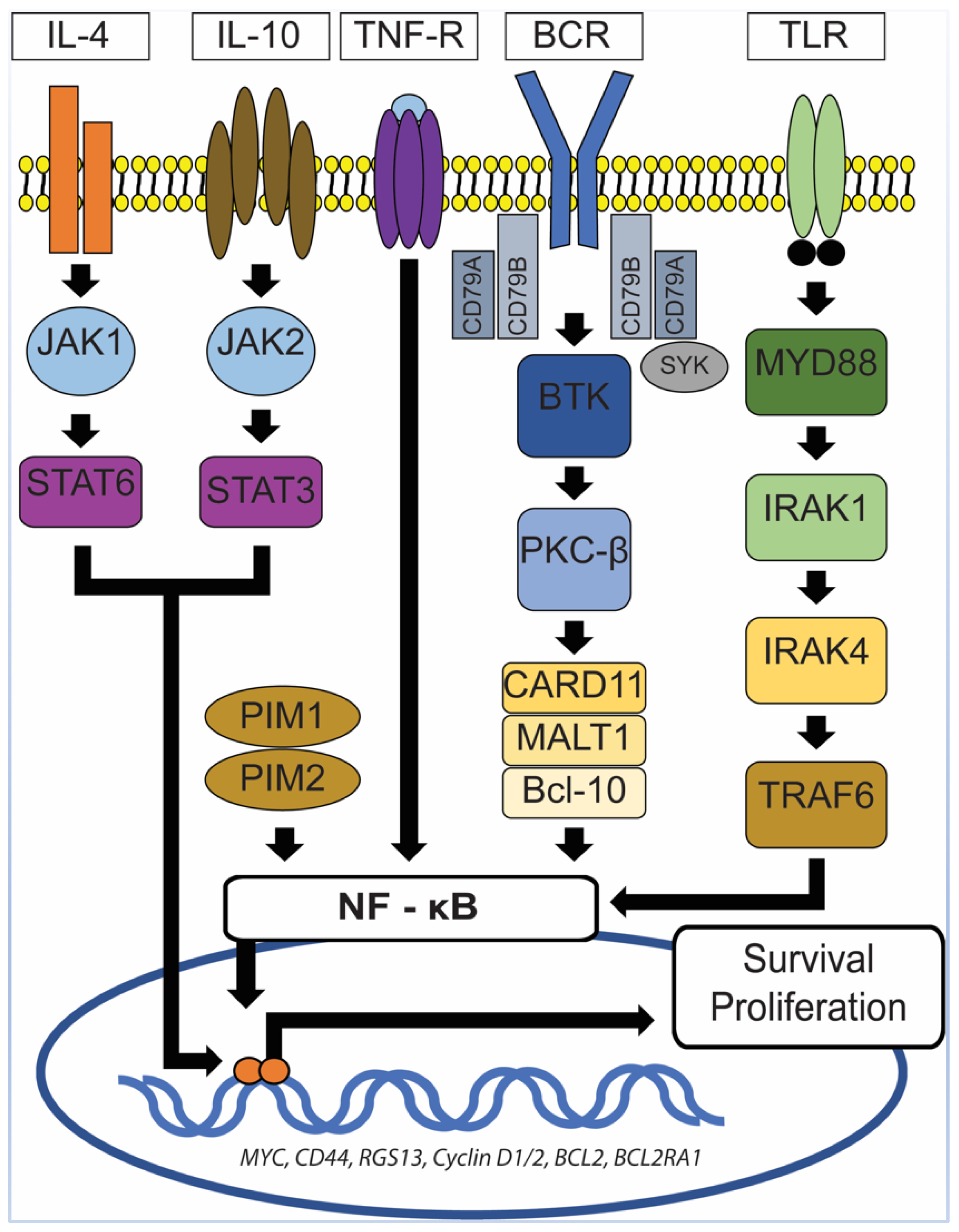{kind=link}
{kind=link}
{kind=link}
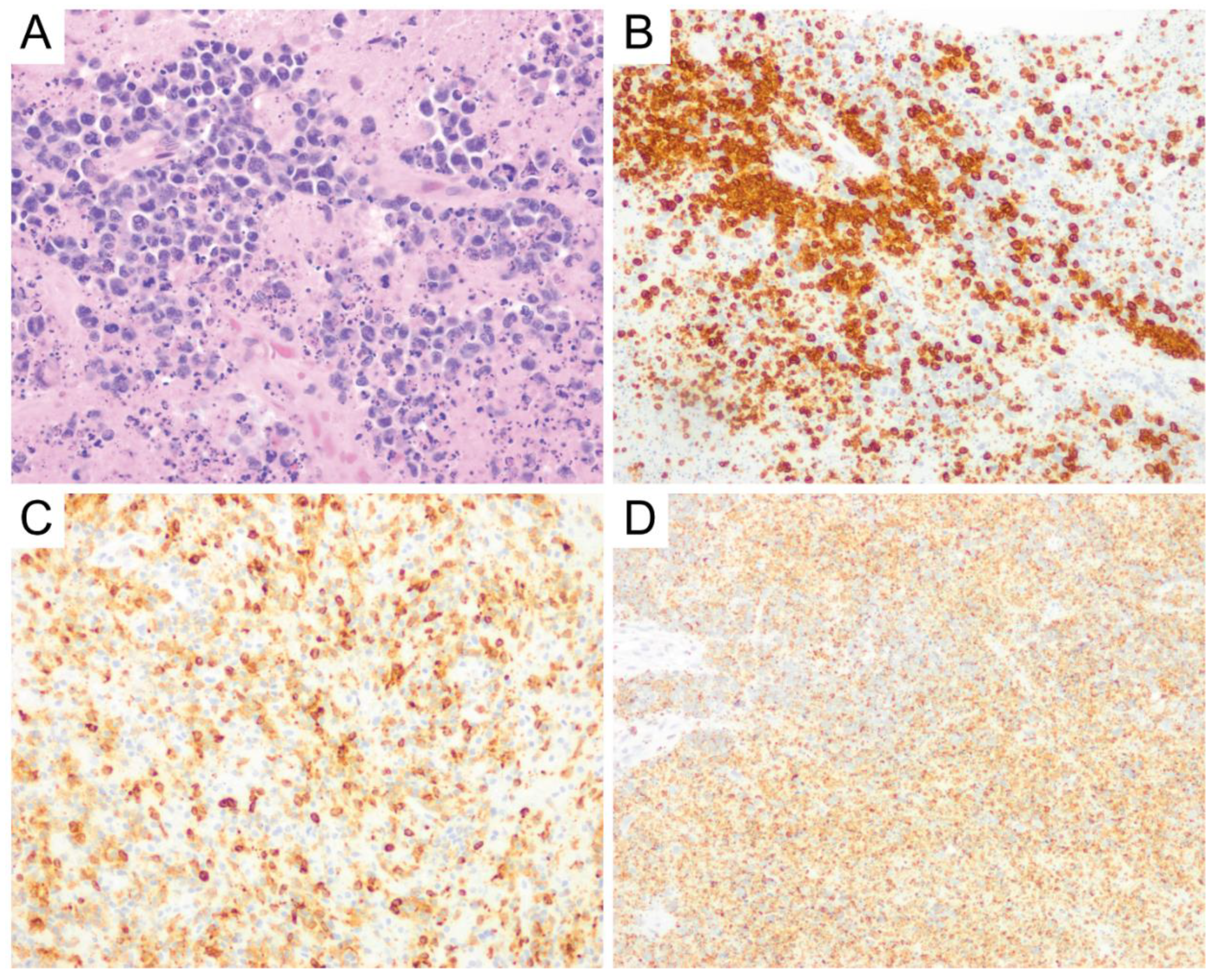{kind=link}
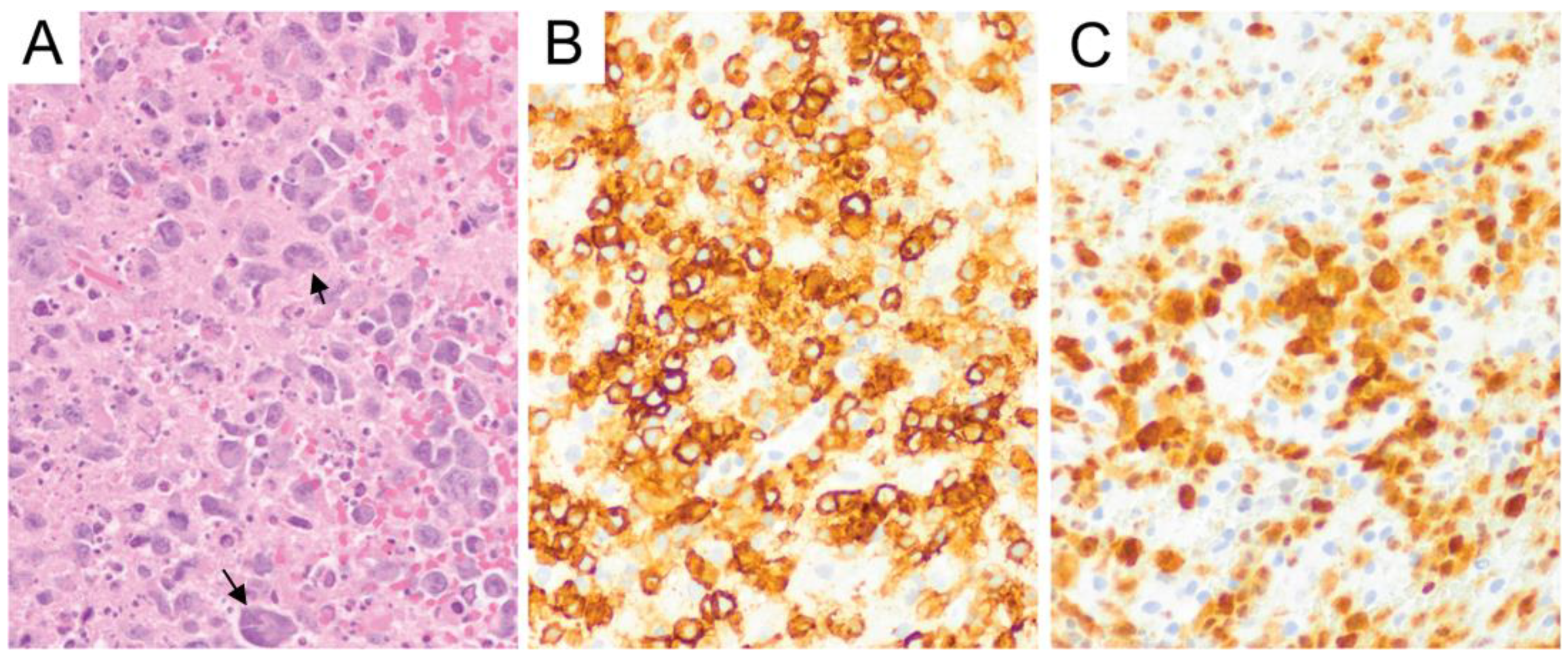{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Clinical Presentation
4. Diagnosis and Staging
5. PCNSL in the Pediatric Population
6. Primary Central Nervous System Diffuse Large B-Cell Lymphoma (PCNS DLBCL)
7. Intravascular Large B-Cell Lymphoma
8. Burkitt Lymphoma
9. Low-Grade B-Cell Lymphomas and T-Cell Lymphomas
10. Dural Marginal Zone Lymphoma (MZL)
11. Peripheral T-Cell Lymphoma, not Otherwise Specified (PTCL, NOS) and Anaplastic Large Cell Lymphoma (ALCL)
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- King, R.L.; Goodlad, J.R.; Calaminici, M.; Dotlic, S.; Montes-Moreno, S.; Oschlies, I.; Ponzoni, M.; Traverse-Glehen, A.; Ott, G.; Ferry, J.A. Lymphomas arising in immune-privileged sites: Insights into biology, diagnosis, and pathogenesis. Virchows Archiv 2019, 476, 647–665. [Google Scholar] [CrossRef]
- Surawicz, T.S.; McCarthy, B.J.; Kupelian, V.; Jukich, P.J.; Bruner, J.M.; Davis, F.G. Descriptive epidemiology of primary brain and CNS tumors: Results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol. 1999, 1, 14–25. [Google Scholar]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18 (Suppl. 5), v1–v75. [Google Scholar] [CrossRef]
- Cote, T.R.; Manns, A.; Hardy, C.R.; Yellin, F.J.; Hartge, P. AIDS/Cancer Study Group Epidemiology of Brain Lymphoma Among People With or Without Acquired Immunodeficiency Syndrome. J. Natl. Cancer Inst. 1996, 88, 675–679. [Google Scholar] [CrossRef]
- Dandachi, D.; Ostrom, Q.T.; Chong, I.; Serpa, J.A.; Giordano, T.P.; Kruchko, C.; Barnholtz-Sloan, J.S.; Fowler, N.; Colen, R.R.; Morón, F.E. Primary central nervous system lymphoma in patients with and without HIV infection: A multicenter study and comparison with U.S national data. Cancer Causes Control. 2019, 30, 477–488. [Google Scholar] [CrossRef]
- Matinella, A.; Lanzafame, M.; Bonometti, M.A.; Gajofatto, A.; Concia, E.; Vento, S.; Monaco, S.; Ferrari, S. Neurological complications of HIV infection in pre-HAART and HAART era: A retrospective study. J. Neurol. 2015, 262, 1317–1327. [Google Scholar] [CrossRef]
- Gopal, S.; Patel, M.R.; Yanik, E.L.; Cole, S.R.; Achenbach, C.J.; Napravnik, S.; Burkholder, G.A.; Reid, E.G.; Rodriguez, B.; Deeks, S.G.; et al. Temporal Trends in Presentation and Survival for HIV-Associated Lymphoma in the Antiretroviral Therapy Era. J. Natl. Cancer Inst. 2013, 105, 1221–1229. [Google Scholar] [CrossRef]
- Bataille, B.; Delwail, V.; Menet, E.; Vandermarcq, P.; Ingrand, P.; Wager, M.; Guy, G.; Lapierre, F. Primary intracerebral malignant lymphoma: Report of 248 cases. J. Neurosurg. 2000, 92, 261–266. [Google Scholar] [CrossRef]
- Grommes, C.; Rubenstein, J.L.; DeAngelis, L.M.; Ferreri, A.J.M.; Batchelor, T.T. Comprehensive approach to diagnosis and treatment of newly diagnosed primary CNS lymphoma. Neuro Oncol. 2019, 21, 296–305. [Google Scholar] [CrossRef]
- Grisariu, S.; Avni, B.; Batchelor, T.T.; Bent, M.J.V.D.; Bokstein, F.; Schiff, D.; Kuittinen, O.; Chamberlain, M.C.; Roth, P.; Nemets, A.; et al. Neurolymphomatosis: An International Primary CNS Lymphoma Collaborative Group report. Blood 2010, 115, 5005–5011. [Google Scholar] [CrossRef]
- Grimm, S.A.; Pulido, J.S.; Jahnke, K.; Schiff, D.; Hall, A.J.; Shenkier, T.N.; Siegal, T.; Doolittle, N.D.; Batchelor, T.T.; Herrlinger, U.; et al. Primary intraocular lymphoma: An International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann. Oncol. 2007, 18, 1851–1855. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, Revised, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Abrey, L.E.; Batchelor, T.T.; Ferreri, A.J.; Gospodarowicz, M.; Pulczynski, E.J.; Zucca, E.; Smith, J.R.; Korfel, A.; Soussain, C.; DeAngelis, L.M.; et al. Report of an International Workshop to Standardize Baseline Evaluation and Response Criteria for Primary CNS Lymphoma. J. Clin. Oncol. 2005, 23, 5034–5043. [Google Scholar] [CrossRef]
- Booman, M.; Douwes, J.; Legdeur, M.-C.; Van Baarlen, J.; Schuuring, E.; Kluin, P.M. From brain to testis: Immune escape and clonal selection in a B cell lymphoma with selective outgrowth in two immune sanctuaries [correction of sanctuariesy]. Haematologica 2007, 92, e69–e71. [Google Scholar] [CrossRef]
- Schmidt, S.; Rainer, J.; Ploner, C.; Presul, E.; Riml, S.; Kofler, R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: Molecular mechanisms and clinical relevance. Cell Death Differ. 2004, 11, S45–S55. [Google Scholar] [CrossRef]
- Geppert, M.; Ostertag, C.B.; Seitz, G.; Kiessling, M. Glucocorticoid therapy obscures the diagnosis of cerebral lymphoma. Acta Neuropathol. 1990, 80, 629–634. [Google Scholar] [CrossRef]
- Barrantes-Freer, A.; Engel, A.S.; Rodríguez-Villagra, O.A.; Winkler, A.; Bergmann, M.; Mawrin, C.; Kuempfel, T.; Pellkofer, H.; Metz, I.; Bleckmann, A.; et al. Diagnostic red flags: Steroid-treated malignant CNS lymphoma mimicking autoimmune inflammatory demyelination. Brain Pathol. 2017, 28, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Alakeel, F.; DePalma, L.; Wu, X. 205 The Challenge of Necrotic Tissue for Lymphoma Work-Up. Am. J. Clin. Pathol. 2018, 149, S87–S88. [Google Scholar] [CrossRef]
- Giannini, C.; Dogan, A.; Salomão, D.R. CNS Lymphoma: A Practical Diagnostic Approach. J. Neuropathol. Exp. Neurol. 2014, 73, 478–494. [Google Scholar] [CrossRef]
- Kadan-Lottick, N.S.; Skluzacek, M.C.; Gurney, J.G. Decreasing incidence rates of primary central nervous system lymphoma. Cancer 2002, 95, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Abla, O.; Weitzman, S.; Blay, J.-Y.; O’Neill, B.P.; Abrey, L.E.; Neuwelt, E.; Doolittle, N.D.; Baehring, J.; Pradhan, K.; Martin, S.E.; et al. Primary CNS lymphoma in children and adolescents: A descriptive analysis from the International Primary CNS Lymphoma Collaborative Group (IPCG). Clin. Cancer Res. 2011, 17, 346–352. [Google Scholar] [CrossRef][Green Version]
- Attarbaschi, A.; Abla, O.; Ronceray, L.; Bansil, S.; Bomken, S.; Burkhardt, B.; Ceppi, F.; Chiang, A.K.S.; Dave, H.; Fedorova, A.; et al. Primary central nervous system lymphoma: Initial features, outcome, and late effects in 75 children and adolescents. Blood Adv. 2019, 3, 4291–4297. [Google Scholar] [CrossRef]
- Hochberg, J.; El-Mallawany, N.K.; Abla, O. Adolescent and young adult non-Hodgkin lymphoma. Br. J. Haematol. 2016, 173, 637–650. [Google Scholar] [CrossRef]
- Paulus, W.; Jellinger, K.; Hallas, C.; Ott, G.; Müller-Hermelink, H.K. Human herpesvirus-6 and Epstein-Barr virus genome in primary cerebral lymphomas. Neurology 1993, 43, 1591–1593. [Google Scholar] [CrossRef]
- Deckert, M.; Hans, V.H.; Eis-Hübinger, A.M.; Prinz, M.; Schaller, C.; Van Roost, D.; Aguzzi, A.; Wiestler, O.D.; Deckert, M. Human herpes virus-8 is not associated with primary central nervous system lymphoma in HIV-negative patients. Acta Neuropathol. 2001, 102, 489–495. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Besleaga, R.; Heinsohn, S.; Siebert, R.; Kabisch, H.; Wiestler, O.D.; Deckert, M. Absence of simian virus 40 DNA sequences in primary central nervous system lymphoma in HIV-negative patients. Virchows Archiv 2004, 444, 436–438. [Google Scholar] [CrossRef]
- Murray, J.; Morgello, S. Polyomaviruses and primary central nervous system lymphomas. Neurology 2004, 63, 1299–1301. [Google Scholar] [CrossRef]
- Ponzoni, M.; Berger, F.; Chassagne-Clement, C.; Tinguely, M.; Jouvet, A.; Ferreri, A.J.M.; Dell’Oro, S.; Terreni, M.R.; Doglioni, C.; Weis, J.; et al. Reactive perivascular T-cell infiltrate predicts survival in primary central nervous system B-cell lymphomas. Br. J. Haematol. 2007, 138, 316–323. [Google Scholar] [CrossRef]
- Makino, K.; Nakamura, H.; Shinojima, N.; Kuroda, J.-I.; Yano, S.; Mikami, Y.; Mukasa, A. BCL2 expression is associated with a poor prognosis independent of cellular origin in primary central nervous system diffuse large B-cell lymphoma. J. Neuro-Oncol. 2018, 140, 115–121. [Google Scholar] [CrossRef]
- Camilleri-Broët, S.; Crinière, E.; Broët, P.; Delwail, V.; Mokhtari, K.; Moreau, A.; Kujas, M.; Raphaël, M.; Iraqi, W.; Sautès-Fridman, C.; et al. A uniform activated B-cell–like immunophenotype might explain the poor prognosis of primary central nervous system lymphomas: Analysis of 83 cases. Blood 2005, 107, 190–196. [Google Scholar] [CrossRef]
- Lin, C.-H.; Kuo, K.; Chuang, S.-S.; Kuo, S.-H.; Chang, J.H.; Chang, K.-C.; Hsu, H.-C.; Tien, H.; Cheng, A.-L. Comparison of the Expression and Prognostic Significance of Differentiation Markers between Diffuse Large B-Cell Lymphoma of Central Nervous System Origin and Peripheral Nodal Origin. Clin. Cancer Res. 2006, 12, 1152–1156. [Google Scholar] [CrossRef]
- Hattab, E.M.; Martin, S.E.; Al-Khatib, S.M.; Kupsky, W.J.; Vance, G.H.; Stohler, R.A.; Czader, M.; Al-Abbadi, M.A. Most primary central nervous system diffuse large B-cell lymphomas occurring in immunocompetent individuals belong to the nongerminal center subtype: A retrospective analysis of 31 cases. Mod. Pathol. 2010, 23, 235–243. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Schmitz, R.; Courts, C.; Stenzel, W.; Bechtel, D.; Niedobitek, G.; Blumcke, I.; Reifenberger, G.; Von Deimling, A.; Jungnickel, B.; et al. Absence of Immunoglobulin Class Switch in Primary Lymphomas of the Central Nervous System. Am. J. Pathol. 2005, 166, 1773–1779. [Google Scholar] [CrossRef]
- Brunn, A.; Nagel, I.; Montesinos-Rongen, M.; Klapper, W.; Vater, I.; Paulus, W.; Hans, V.; Blümcke, I.; Weis, J.; Siebert, R.; et al. Frequent triple-hit expression of MYC, BCL2, and BCL6 in primary lymphoma of the central nervous system and absence of a favorable MYClowBCL2low subgroup may underlie the inferior prognosis as compared to systemic diffuse large B cell lymphomas. Acta Neuropathol. 2013, 126, 603–605. [Google Scholar] [CrossRef]
- Gill, K.Z.; Iwamoto, F.; Allen, A.; Hoehn, D.; Murty, V.V.; Alobeid, B.; Bhagat, G. MYC Protein Expression in Primary Diffuse Large B-Cell Lymphoma of the Central Nervous System. PLoS ONE 2014, 9, e114398. [Google Scholar] [CrossRef]
- Cady, F.M.; O’Neill, B.P.; Law, M.E.; Decker, P.A.; Kurtz, D.M.; Giannini, C.; Porter, A.B.; Kurtin, P.J.; Johnston, P.B.; Dogan, A.; et al. Del(6)(q22) and BCL6 Rearrangements in Primary CNS Lymphoma Are Indicators of an Aggressive Clinical Course. J. Clin. Oncol. 2008, 26, 4814–4819. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Zühlke-Jenisch, R.; Gesk, S.; Martín-Subero, J.I.; Schaller, C.; Van Roost, D.; Wiestler, O.D.; Deckert, M.; Siebert, R. Interphase Cytogenetic Analysis of Lymphoma-Associated Chromosomal Breakpoints in Primary Diffuse Large B-Cell Lymphomas of the Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 926–933. [Google Scholar] [CrossRef]
- Shi, Q.-Y.; Feng, X.; Bao, W.; Ma, J.; Lv, J.-H.; Wang, X.; Rao, Q.; Shi, Q.-L. MYC/BCL2 Co-Expression Is a Stronger Prognostic Factor Compared With the Cell-of-Origin Classification in Primary CNS DLBCL. J. Neuropathol. Exp. Neurol. 2017, 76, 942–948. [Google Scholar] [CrossRef]
- Kim, S.; Nam, S.J.; Kwon, D.; Kim, H.; Lee, E.; Kim, T.M.; Heo, D.S.; Park, S.H.; Kim, C.W.; Jeon, Y.K. MYC and BCL2 overexpression is associated with a higher class of Memorial Sloan-Kettering Cancer Center prognostic model and poor clinical outcome in primary diffuse large B-cell lymphoma of the central nervous system. BMC Cancer 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Tapia, G.; Baptista, M.-J.; Muñoz-Marmol, A.-M.; Gaafar, A.; Puente-Pomposo, M.; Garcia, O.; Marginet-Flinch, R.; Sanz, C.; Navarro, J.-T.; Sancho, J.-M.; et al. MYC protein expression is associated with poor prognosis in primary diffuse large B-cell lymphoma of the central nervous system. APMIS 2015, 123, 596–603. [Google Scholar] [CrossRef]
- Fukumura, K.; Kawazu, M.; Kojima, S.; Ueno, T.; Sai, E.; Soda, M.; Ueda, H.; Yasuda, T.; Yamaguchi, H.; Lee, J.; et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016, 131, 865–875. [Google Scholar] [CrossRef]
- Son, S.-M.; Ha, S.-Y.; Yoo, H.-Y.; Oh, D.; Kim, S.J.; Kim, W.-S.; Ko, Y.-H. Prognostic impact of MYC protein expression in central nervous system diffuse large B-cell lymphoma: Comparison with MYC rearrangement and MYC mRNA expression. Mod. Pathol. 2016, 30, 4–14. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Liu, Y.; Liu, Z.; Cui, Q.; Ji, N.; Sun, S.; Wang, B.; Wang, Y.; Sun, X.; et al. Immunohistochemical profile and prognostic significance in primary central nervous system lymphoma: Analysis of 89 cases. Oncol. Lett. 2017, 14, 5505–5512. [Google Scholar] [CrossRef]
- Guo, S.; Bai, Q.; Rohr, J.M.; Wang, Y.; Liu, Y.; Zeng, K.; Yu, K.; Zhang, X.; Wang, Z. Clinicopathological features of primary diffuse large B-cell lymphoma of the central nervous system-strong EZH2 expression implying diagnostic and therapeutic implication. APMIS 2016, 124, 1054–1062. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Küppers, R.; Schlüter, D.; Spieker, T.; Van Roost, D.; Schaller, C.; Reifenberger, G.; Wiestler, O.D.; Deckert-Schlüter, M. Primary Central Nervous System Lymphomas Are Derived from Germinal-Center B Cells and Show a Preferential Usage of the V4–34 Gene Segment. Am. J. Pathol. 1999, 155, 2077–2086. [Google Scholar] [CrossRef]
- Sharathkumar, B.; Jon, D.W. Primary Central Nervous System Lymphoma. Arch. Pathol. Lab. Med. 2008, 132, 1830–1834. [Google Scholar]
- Rubenstein, J.L.; Fridlyand, J.; Shen, A.; Aldape, K.; Ginzinger, D.; Batchelor, T.; Treseler, P.; Berger, M.; McDermott, M.; Prados, M.; et al. Gene expression and angiotropism in primary CNS lymphoma. Blood 2006, 107, 3716–3723. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Godlewska, E.; Brunn, A.; Wiestler, O.D.; Siebert, R.; Deckert, M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011, 122, 791–792. [Google Scholar] [CrossRef]
- Schwindt, H.; Vater, I.; Kreuz, M.; Montesinos-Rongen, M.; Brunn, A.; Richter, J.; Gesk, S.; Ammerpohl, O.; Wiestler, O.D.; Hasenclever, D.; et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia 2009, 23, 1875–1884. [Google Scholar] [CrossRef][Green Version]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Dognini, G.P.; Campo, E.; Willemze, R.; Seymour, J.F.; Bairey, O.; Martelli, M.; De Renz, A.O.; Doglioni, C.; Montalbán, C.; et al. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Haematologica 2007, 92, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.E.; Kim, W.S.; Kim, S.J. Asian variant of intravascular large B-cell lymphoma: A comparison of clinical features based on involvement of the central nervous system. Korean J. Intern. Med. 2020, 35, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Tahsili-Fahadan, P.; Rashidi, A.; Cimino, P.J.; Bucelli, R.C.; Keyrouz, S.G. Neurologic manifestations of intravascular large B-cell lymphoma. Neurol. Clin. Pr. 2015, 6, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Matsue, K.; Abe, Y.; Narita, K.; Kobayashi, H.; Kitadate, A.; Takeuchi, M.; Miura, D.; Takeuchi, K. Diagnosis of intravascular large B cell lymphoma: Novel insights into clinicopathological features from 42 patients at a single institution over 20 years. Br. J. Haematol. 2019, 187, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Murase, T.; Matsue, K.; Okamoto, M.; Ichikawa, N.; Tsukamoto, N.; Niitsu, N.; Miwa, H.; Asaoku, H.; Kosugi, H.; et al. Central nervous system involvement in intravascular large B-cell lymphoma: A retrospective analysis of 109 patients. Cancer Sci. 2010, 101, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Arrigoni, G.; Gould, V.E.; Del Curto, B.; Maggioni, M.; Scapinello, A.; Paolino, S.; Cassisa, A.; Patriarca, C. Lack of CD 29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol. 2000, 31, 220–226. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.H.; Cleton-Jansen, A.-M.; Somers, S.F.; Veelken, H.; Van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef]
- Bower, K.; Shah, N. Primary CNS Burkitt Lymphoma: A Case Report of a 55-Year-Old Cerebral Palsy Patient. Case Rep. Oncol. Med. 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Pesin, N.; Lam, C.; Margolin, E. Central Nervous System Burkitt Lymphoma Presenting as Atypical Guillain-Barre Syndrome. Can. J. Neurol. Sci. 2019, 47, 145–147. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sano, T.; Ii, K.; Hizawa, K.; Li, K. Primary Burkitt-type lymphoma of the central nervous system. Acta Neuropathol. 1984, 64, 12–14. [Google Scholar] [CrossRef]
- Leucci, E.; Cocco, M.; Onnis, A.; De Falco, G.; Van Cleef, P.; Bellan, C.; Van Rijk, A.; Nyagol, J.; Byakika, B.; Lazzi, S.; et al. MYC translocation-negative classical Burkitt lymphoma cases: An alternative pathogenetic mechanism involving miRNA deregulation. J. Pathol. 2008, 216, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kretzmer, H.; Project, I.M.-S.; Bernhart, S.H.; Wang, W.; Haake, A.; Weniger, M.A.; Bergmann, A.K.; Betts, M.J.; Carrillo-De-Santa-Pau, E.; Doose, G.; et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 2015, 47, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.D.; Li, G.; Zhang, J.; Miles, R.R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Project, T.I.M.-S.; Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Giulino-Roth, L.; Wang, K.; Macdonald, T.Y.; Mathew, S.; Tam, Y.; Cronin, M.T.; Palmer, G.; Lucena-Silva, N.; Pedrosa, F.; Pedrosa, M.; et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 2012, 120, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Wagener, R.; Aukema, S.M.; Schlesner, M.; Haake, A.; Burkhardt, B.; Claviez, A.; Drexler, H.G.; Hummel, M.; Kreuz, M.; Loeffler, M.; et al. ThePCBP1gene encoding poly(rc) binding protein i is recurrently mutated in Burkitt lymphoma. Genes Chromosom. Cancer 2015, 54, 555–564. [Google Scholar] [CrossRef]
- Jahnke, K.; Korfel, A.; O’Neill, B.P.; Blay, J.-Y.; Abrey, L.E.; Martus, P.; Poortmans, P.M.P.; Shenkier, T.N.; Batchelor, T.T.; Neuwelt, E.A.; et al. International study on low-grade primary central nervous system lymphoma. Ann. Neurol. 2006, 59, 755–762. [Google Scholar] [CrossRef]
- Papanicolau-Sengos, A.; Wang-Rodriguez, J.; Wang, H.-Y.; Lee, R.R.; Wong, A.; Hansen, L.A.; Mahooti, S.; Rashidi, H.H. Rare case of a primary non-dural central nervous system low grade B-cell lymphoma and literature review. Int. J. Clin. Exp. Pathol. 2012, 5, 89–95. [Google Scholar]
- Nomani, L.; Cotta, C.V.; Hsi, E.D.; A Ferry, J.; Cook, J.R. Extranodal Marginal Zone Lymphoma of the Central Nervous System Includes Parenchymal-Based Cases With Characteristic Features. Am. J. Clin. Pathol. 2020, 154, 124–132. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Kuzu, I.; Dogan, A.; Dirnhofer, S.; Chan, J.K.C.; Sander, B.; Ott, G.; Xerri, L.; Quintanilla-Martinez, L.; Campo, E. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Archiv 2016, 468, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, K.A.; Jobanputra, V.; Iwamoto, F.; Jain, P.; Chen, J.; Cascione, L.; Nahum, O.; Levy, B.; Xie, Y.; Khattar, P.; et al. The genetic landscape of dural marginal zone lymphomas. Oncotarget 2016, 7, 43052–43061. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, D.; Kaldjian, E.P.; Bauserman, S.; Raffeld, M.; Jaffe, E.S. Primary low-grade B-cell lymphoma of the dura: A mucosa associated lymphoid tissue-type lymphoma. Am. J. Surg. Pathol. 1997, 21, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.; Giannini, C.; Judkins, A.R.; Schwalb, J.M.; Burack, R.; O’Neill, B.P.; Yachnis, A.T.; Burger, P.C.; Scheithauer, B.W.; Perry, A. Clinicopathologic and Genetic Profile of Intracranial Marginal Zone Lymphoma: A Primary Low-Grade CNS Lymphoma That Mimics Meningioma. J. Clin. Oncol. 2005, 23, 5718–5727. [Google Scholar] [CrossRef]
- Matmati, K.S.; Matmati, N.; Hannun, Y.A.; Rumboldt, Z.; Patel, S.; Lazarchick, J.; Stuart, R.; Giglio, P. Dural MALT lymphoma with disseminated disease. Hematol. Rep. 2010, 2, 10. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; DeAngelis, L.M.; Abrey, L.E. Primary dural lymphomas: A clinicopathologic study of treatment and outcome in eight patients. Neurol. 2006, 66, 1763–1765. [Google Scholar] [CrossRef]
- De La Fuente, M.I.; Haggiagi, A.; Moul, A.; Young, R.J.; Sidani, C.; Markoe, A.; Vega, F.; DeAngelis, L.M.; Lossos, I.S. Marginal zone dural lymphoma: The Memorial Sloan Kettering Cancer Center and University of Miami experiences. Leuk. Lymphoma 2016, 58, 882–888. [Google Scholar] [CrossRef]
- Venkataraman, G.; Rizzo, K.A.; Chavez, J.J.; Streubel, B.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S. Marginal zone lymphomas involving meningeal dura: Possible link to IgG4-related diseases. Mod. Pathol. 2011, 24, 355–366. [Google Scholar] [CrossRef]
- Hayabuchi, N.; Shibamoto, Y.; Onizuka, Y. Primary central nervous system lymphoma in japan: A nationwide survey. Int. J. Radiat. Oncol. 1999, 44, 265–272. [Google Scholar] [CrossRef]
- Lim, T.; Kim, S.J.; Kim, K.; Lee, J.-I.; Lim, D.H.; Lee, D.J.; Baek, K.K.; Lee, H.Y.; Han, B.; Uhm, J.E.; et al. Primary CNS lymphoma other than DLBCL: A descriptive analysis of clinical features and treatment outcomes. Ann. Hematol. 2011, 90, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Shenkier, T.N.; Blay, J.-Y.; O’Neill, B.P.; Poortmans, P.; Thiel, E.; Jahnke, K.; Abrey, L.E.; Neuwelt, E.; Tsang, R.; Batchelor, T.; et al. Primary CNS Lymphoma of T-Cell Origin: A Descriptive Analysis From the International Primary CNS Lymphoma Collaborative Group. J. Clin. Oncol. 2005, 23, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Villegas, E.; Villà, S.; López-Guillermo, A.; Petit, J.; Ribalta, T.; Graus, F. Primary central nervous system lymphoma of T-cell origin: Description of two cases and review of the literature. J. Neuro-Oncol. 1997, 34, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.P.; Nicolae, A.; Meeker, H.; Raffeld, M.; Xi, L.; Jegalian, A.G.; Miller, C.D.; Pittaluga, S.; Jaffe, E.S. Primary CNS T-cell Lymphomas: A Clinical, Morphologic, Immunophenotypic, and Molecular Analysis. Am. J. Surg. Pathol. 2015, 39, 1719. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauw, M.I.S.; Lucas, C.-H.G.; Ohgami, R.S.; Wen, K.W. Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features. Diagnostics 2020, 10, 1076. https://doi.org/10.3390/diagnostics10121076
Lauw MIS, Lucas C-HG, Ohgami RS, Wen KW. Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features. Diagnostics. 2020; 10(12):1076. https://doi.org/10.3390/diagnostics10121076
Chicago/Turabian StyleLauw, Marietya I. S., Calixto-Hope G. Lucas, Robert S. Ohgami, and Kwun Wah Wen. 2020. "Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features" Diagnostics 10, no. 12: 1076. https://doi.org/10.3390/diagnostics10121076
APA StyleLauw, M. I. S., Lucas, C.-H. G., Ohgami, R. S., & Wen, K. W. (2020). Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features. Diagnostics, 10(12), 1076. https://doi.org/10.3390/diagnostics10121076