A Novel DSP Truncating Variant in a Family with Episodic Myocardial Injury in the Course of Arrhythmogenic Cardiomyopathy—A Possible Role of a Low Penetrance NLRP3 Variant

 , , , and
, , , and 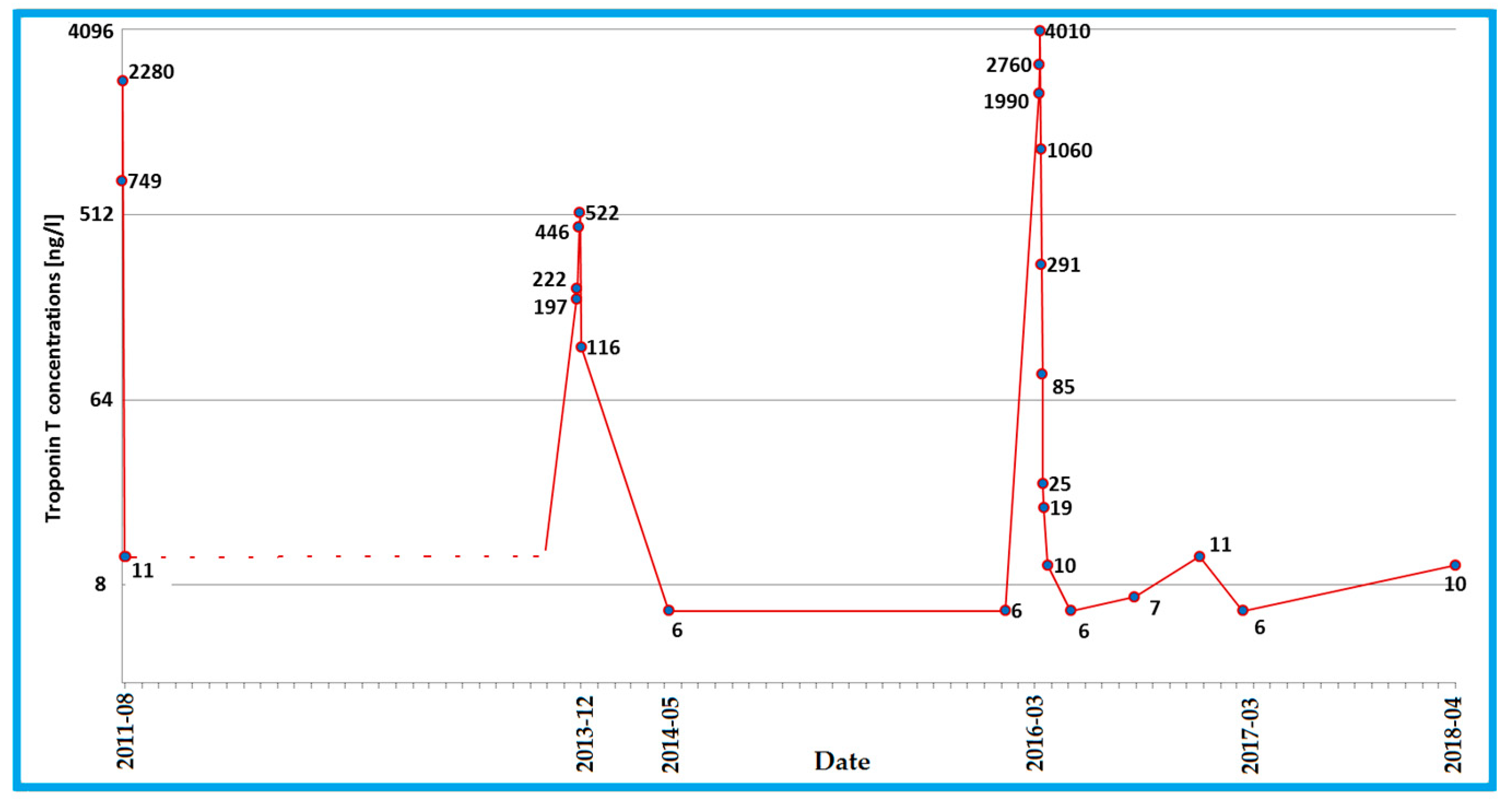{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
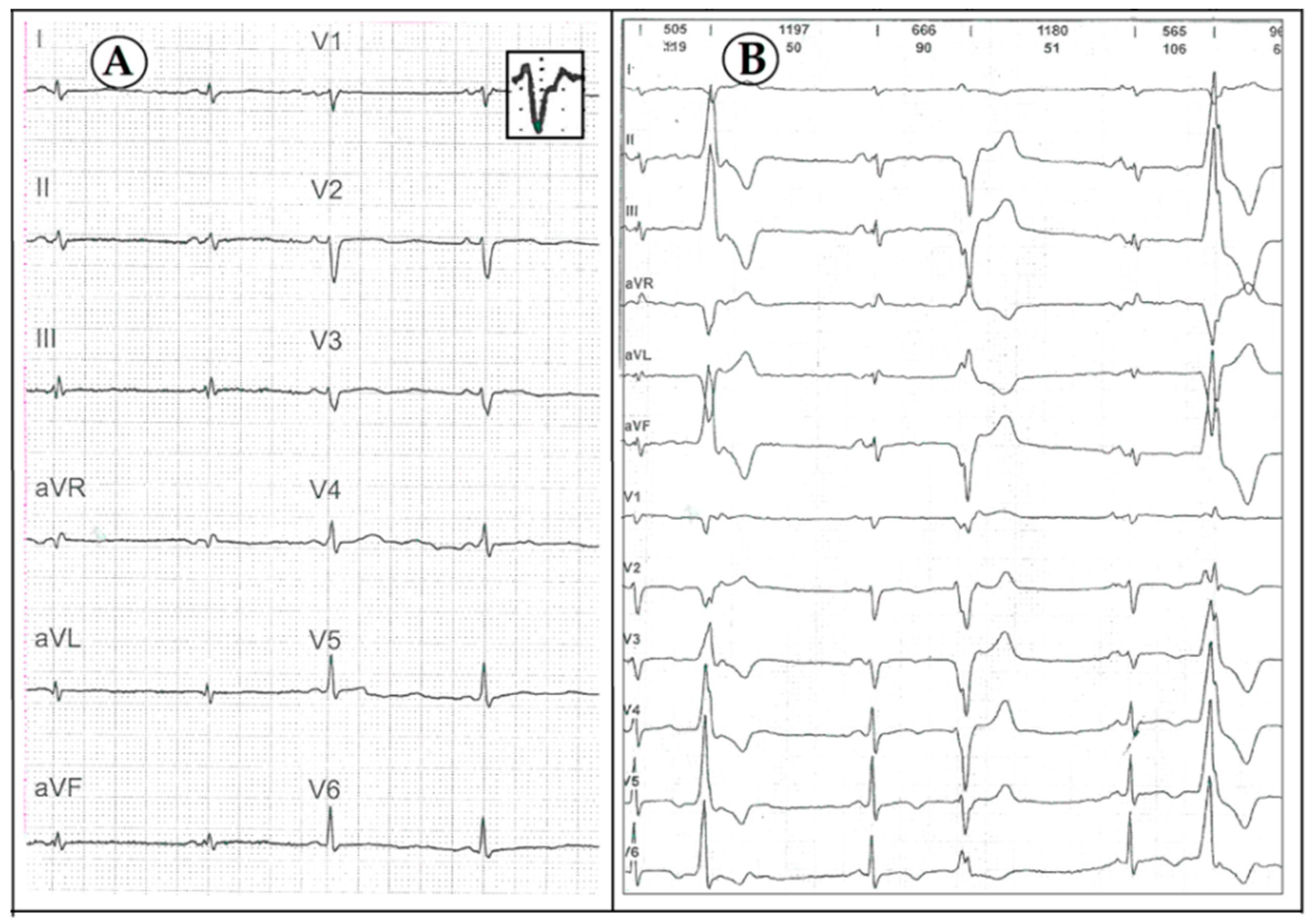
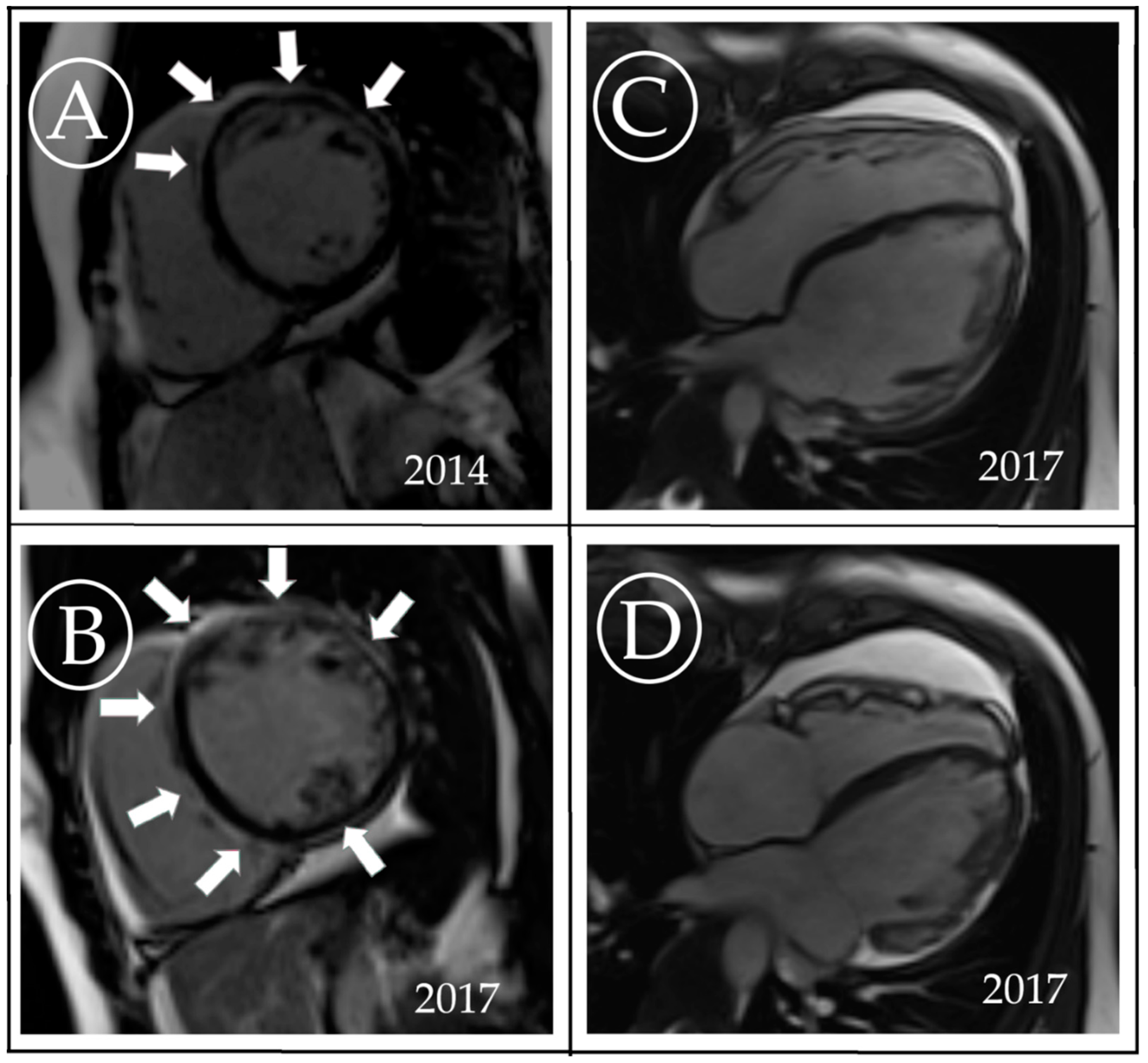
3. Case Description
4. Discussion
4.1. DSP—Novel Variant
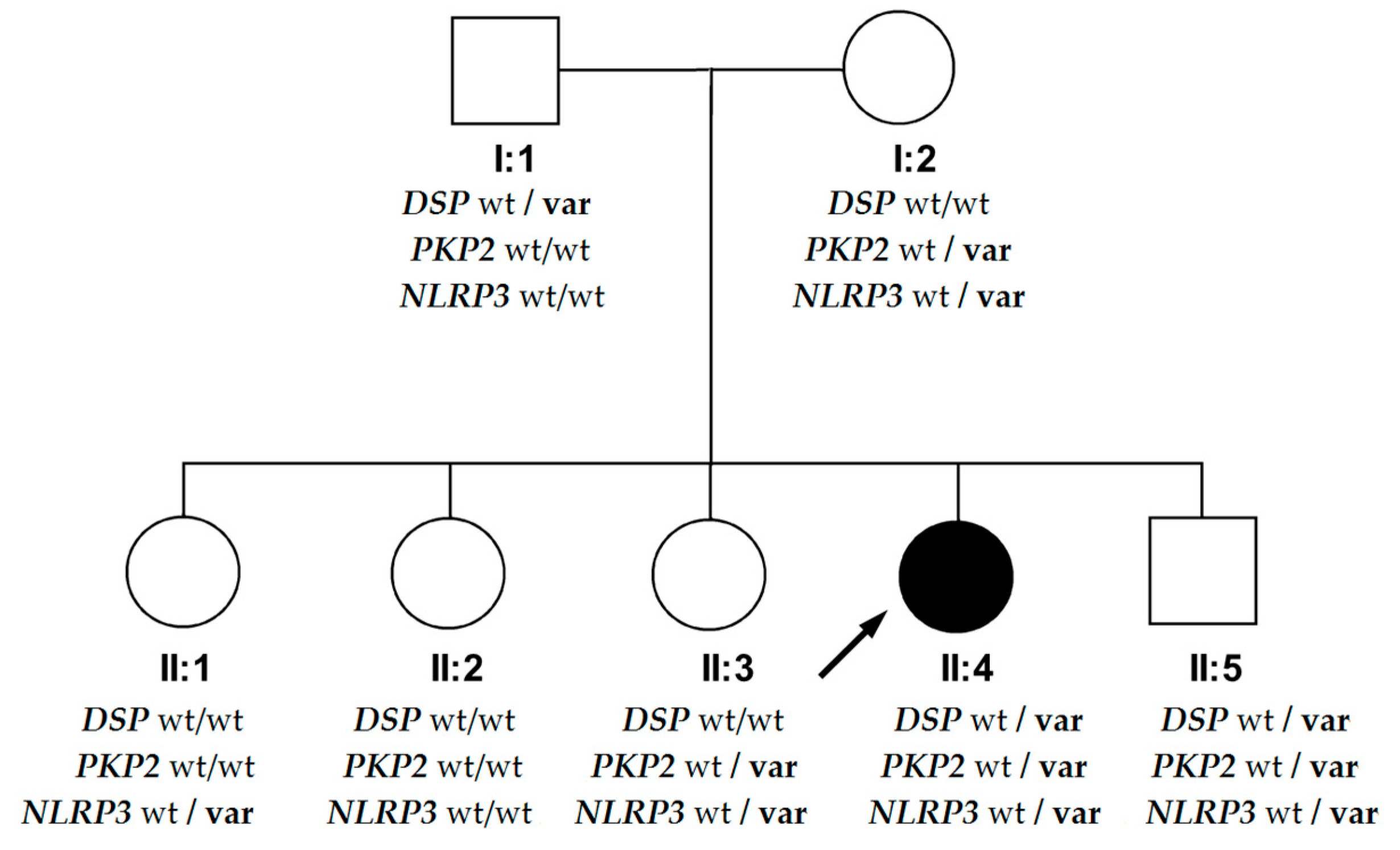
4.2. Phenotypic Differences in the DSP Variant’s Carriers within the Family
5. Study Limitations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef]
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Rivera Sorensen, C.; Sheppard, M.N.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Syrris, P.; Salas, C.; Evans, A.; Mirelis, J.G.; Cobo-Marcos, M.; Vilches, C.; Bornstein, B.; Segovia, J.; Alonso-Pulpon, L.; et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: A clinicopathological study. Heart 2011, 97, 1744–1752. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Stępień-Wojno, M.; Franaszczyk, M.; Bodalski, R.; Śpiewak, M.; Baranowski, R.S.; Grzybowski, J.; Płoski, R.; Bilińska, Z.T. A different background of arrhythmia in siblings with a positive family history of sudden death at young age. Ann. Noninvasive Electrocardiol. 2019, e12707. [Google Scholar]
- Chmielewski, P.; Michalak, E.; Kowalik, I.; Franaszczyk, M.; Sobieszczanska-Malek, M.; Truszkowska, G.; Stepien-Wojno, M.; Biernacka, E.K.; Foss-Nieradko, B.; Lewandowski, M.; et al. Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers? J. Clin. Med. 2020, 9, 1443. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef]
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Béziau, D.M.; Marsman, R.F.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal ectopic Purkinje-related premature contractions: A new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156. [Google Scholar] [CrossRef]
- Zakrzewska-Koperska, J.; Franaszczyk, M.; Bilińska, Z.; Truszkowska, G.; Karczmarz, M.; Szumowski, Ł.; Zieliński, T.; Płoski, R.; Bilińska, M. Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: A case report. BMC Med. Genet. 2018, 19, 94. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Truszkowska, G.T.; Bilińska, Z.T.; Kosińska, J.; Śleszycka, J.; Rydzanicz, M.; Sobieszczańska-Małek, M.; Franaszczyk, M.; Bilińska, M.; Stawiński, P.; Michalak, E.; et al. A study in Polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (PLN) mutations at amino acid position 9 and low penetrance of heterozygous null PLN mutations. BMC Med. Genet. 2015, 16, 21. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Truszkowska, G.; Chmielewski, P.; Rydzanicz, M.; Kosinska, J.; Rywik, T.; Biernacka, A.; Spiewak, M.; Kostrzewa, G.; Stepien-Wojno, M.; et al. Analysis of De Novo Mutations in Sporadic Cardiomyopathies Emphasizes Their Clinical Relevance and Points to Novel Candidate Genes. J. Clin. Med. 2020, 9, 370. [Google Scholar] [CrossRef]
- Stępień-Wojno, M.; Ponińska, J.; Rydzanicz, M.; Bilińska, M.; Truszkowska, G.; Baranowski, R.; Lutyńska, A.; Biernacka, E.K.; Stępińska, J.; Kowalik, I.; et al. Sudden cardiac arrest in patients without overt heart disease: A limited value of next generation sequencing. Pol. Arch. Intern. Med. 2018, 128, 721–730. [Google Scholar]
- Foss-Nieradko, B.; Franaszczyk, M.; Śpiewak, M.; Oręziak, A.; Płoski, R.; Bilińska, Z.T. Novel truncating desmoplakin mutation as a potential cause of sudden cardiac death in a family. Pol. Arch. Med. Med. 2016, 126, 704–707. [Google Scholar] [CrossRef]
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- Mahoney, M.G.; Sadowski, S.; Brennan, D.; Pikander, P.; Saukko, P.; Wahl, J.; Aho, H.; Heikinheimo, K.; Bruckner-Tuderman, L.; Fertala, A.; et al. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J. Invest. Dermatol. 2010, 130, 968–978. [Google Scholar] [CrossRef]
- Bari, O.; Skillman, S.; Lah, M.D.; Haggstrom, A.N. Compound heterozygous mutations in desmoplakin associated with skin fragility, follicular hyperkeratosis, alopecia, and nail dystrophy. Pediatr. Dermatol. 2018, 35, e218–e220. [Google Scholar] [CrossRef]
- Surmacz, R.; Franaszczyk, M.; Pyda, M.; Płoski, R.; Bilińska, Z.T.; Bobkowski, W. Autosomal recessive transmission of familial nonsyndromic dilated cardiomyopathy due to compound desmoplakin gene mutations. Pol. Arch. Intern. Med. 2018, 128, 785–787. [Google Scholar]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Poller, W.; Haas, J.; Klingel, K.; Kühnisch, J.; Gast, M.; Kaya, Z.; Escher, F.; Kayvanpour, E.; Degener, F.; Opgen-Rhein, B.; et al. Familial Recurrent Myocarditis Triggered by Exercise in Patients With a Truncating Variant of the Desmoplakin Gene. J. Am. Heart Assoc. 2020, 9, e015289. [Google Scholar] [CrossRef]
- Kissopoulou, A.; Fernlund, E.; Holmgren, C.; Isaksson, E.; Karlsson, J.E.; Green, H.; Jonasson, J.; Ellegård, R.; Årstrand, H.K.; Svensson, A.; et al. Monozygotic twins with myocarditis and a novel likely pathogenic desmoplakin gene variant. ESC Heart Fail. 2020, 7, 1210–1216. [Google Scholar] [CrossRef]
- Patriki, D.; Gresser, E.; Manka, R.; Emmert, M.Y.; Lüscher, T.F.; Heidecker, B. Approximation of the Incidence of Myocarditis by Systematic Screening With Cardiac Magnetic Resonance Imaging. JACC Heart Fail. 2018, 6, 573–579. [Google Scholar] [CrossRef]
- Kufer, T.A.; Banks, D.J.; Philpott, D.J. Innate immune sensing of microbes by Nod proteins. Ann. N. Y. Acad. Sci. 2006, 1072, 19–27. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Torres, A.; Brownstein, C.A.; Tembulkar, S.K.; Graber, K.; Genetti, C.; Kleiman, R.J.; Sweadner, K.J.; Mavros, C.; Liu, K.X.; Smedemark-Margulies, N.; et al. De novo ATP1A3 and compound heterozygous NLRP3 mutations in a child with autism spectrum disorder, episodic fatigue and somnolence, and muckle-wells syndrome. Mol. Genet. Metab. Rep. 2018, 16, 23–29. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Verma, D.; Endres, T.; Broderick, L.; de Jesus, A.A.; Hofer, F.; Blank, N.; Krause, K.; Rietschel, C.; Horneff, G.; et al. Clinical and Molecular Phenotypes of Low-Penetrance Variants of NLRP3: Diagnostic and Therapeutic Challenges. Arthritis Rheumatol. 2017, 69, 2233–2240. [Google Scholar] [CrossRef]
- Rieber, N.; Gavrilov, A.; Hofer, L.; Singh, A.; Öz, H.; Endres, T.; Schäfer, I.; Handgretinger, R.; Hartl, D.; Kuemmerle-Deschner, J. A functional inflammasome activation assay differentiates patients with pathogenic NLRP3 mutations and symptomatic patients with low penetrance variants. Clin. Immunol. 2015, 157, 56–64. [Google Scholar] [CrossRef]
- Blomgran, R.; Patcha Brodin, V.; Verma, D.; Bergström, I.; Söderkvist, P.; Sjöwall, C.; Eriksson, P.; Lerm, M.; Stendahl, O.; Särndahl, E. Common genetic variations in the NALP3 inflammasome are associated with delayed apoptosis of human neutrophils. PLoS ONE 2012, 7, e31326. [Google Scholar] [CrossRef]
- Ploski, R.; Pollak, A.; Müller, S.; Franaszczyk, M.; Michalak, E.; Kosinska, J.; Stawinski, P.; Spiewak, M.; Seggewiss, H.; Bilinska, Z.T. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ. Res. 2014, 114, e2–e5. [Google Scholar] [CrossRef]
- Uzumcu, A.; Norgett, E.E.; Dindar, A.; Uyguner, O.; Nisli, K.; Kayserili, H.; Sahin, S.E.; Dupont, E.; Severs, N.J.; Leigh, I.M.; et al. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J. Med. Genet. 2006, 43, e05. [Google Scholar] [CrossRef]
- Cabral, R.M.; Wan, H.; Cole, C.L.; Abrams, D.J.; Kelsell, D.P.; South, A.P. Identification and characterization of DSPIa, a novel isoform of human desmoplakin. Cell Tissue Res. 2010, 341, 121–129. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef]
- Aróstegui, J.I.; Aldea, A.; Modesto, C.; Rua, M.J.; Argüelles, F.; González-Enseñat, M.A.; Ramos, E.; Rius, J.; Plaza, S.; Vives, J.; et al. Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/PYPAF1/NALP3 gene. Arthritis Rheumatol. 2004, 50, 4045–4050. [Google Scholar]
- Kanneganti, T.D.; Ozören, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Rae, W.; Ward, D.; Mattocks, C.; Pengelly, R.J.; Eren, E.; Patel, S.V.; Faust, S.N.; Hunt, D.; Williams, A.P. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin. Genet. 2018, 93, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, M.H.; van de Vosse, E.; Goldbach-Mansky, R.; Holland, S.M. Impaired cytokine responses in patients with cryopyrin-associated periodic syndrome (CAPS). Clin. Exp. Immunol. 2014, 177, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Groß, C.J.; Wagner, D.; Schlüter, M.; Groß, O.; Kümpfel, T. MCC950 blocks enhanced interleukin-1β production in patients with NLRP3 low penetrance variants. Clin. Immunol. 2019, 203, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Lowe, P.; Felix, S.D.; Rordorf, C.; Leslie, K.; Madhoo, S.; Wittkowski, H.; Bek, S.; Hartmann, N.; Bosset, S.; et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J. Exp. Med. 2009, 206, 1029–1036. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Putnam, C.D.; Remmers, E.F.; Mueller, J.L.; Le, J.; Kolodner, R.D.; Moak, Z.; Chuang, M.; Austin, F.; Goldbach-Mansky, R.; et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheumatol. 2007, 56, 1273–1285. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmielewski, P.; Truszkowska, G.T.; Kukla, P.; Zakrzewska-Koperska, J.; Śpiewak, M.; Stępień-Wojno, M.; Bilińska, M.; Lutyńska, A.; Płoski, R.; Bilińska, Z.T. A Novel DSP Truncating Variant in a Family with Episodic Myocardial Injury in the Course of Arrhythmogenic Cardiomyopathy—A Possible Role of a Low Penetrance NLRP3 Variant. Diagnostics 2020, 10, 955. https://doi.org/10.3390/diagnostics10110955
Chmielewski P, Truszkowska GT, Kukla P, Zakrzewska-Koperska J, Śpiewak M, Stępień-Wojno M, Bilińska M, Lutyńska A, Płoski R, Bilińska ZT. A Novel DSP Truncating Variant in a Family with Episodic Myocardial Injury in the Course of Arrhythmogenic Cardiomyopathy—A Possible Role of a Low Penetrance NLRP3 Variant. Diagnostics. 2020; 10(11):955. https://doi.org/10.3390/diagnostics10110955
Chicago/Turabian StyleChmielewski, Przemysław, Grażyna T. Truszkowska, Piotr Kukla, Joanna Zakrzewska-Koperska, Mateusz Śpiewak, Małgorzata Stępień-Wojno, Maria Bilińska, Anna Lutyńska, Rafał Płoski, and Zofia T. Bilińska. 2020. "A Novel DSP Truncating Variant in a Family with Episodic Myocardial Injury in the Course of Arrhythmogenic Cardiomyopathy—A Possible Role of a Low Penetrance NLRP3 Variant" Diagnostics 10, no. 11: 955. https://doi.org/10.3390/diagnostics10110955
APA StyleChmielewski, P., Truszkowska, G. T., Kukla, P., Zakrzewska-Koperska, J., Śpiewak, M., Stępień-Wojno, M., Bilińska, M., Lutyńska, A., Płoski, R., & Bilińska, Z. T. (2020). A Novel DSP Truncating Variant in a Family with Episodic Myocardial Injury in the Course of Arrhythmogenic Cardiomyopathy—A Possible Role of a Low Penetrance NLRP3 Variant. Diagnostics, 10(11), 955. https://doi.org/10.3390/diagnostics10110955