Personalization of Aspirin Therapy Ex Vivo in Patients with Atherosclerosis Using Light Transmission Aggregometry


 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Experimental Section
2.1. Ethics Approval
2.2. Patient Selection
2.3. Baseline Measurements
2.4. Blood Sample Collection and Light Transmission Aggregometry
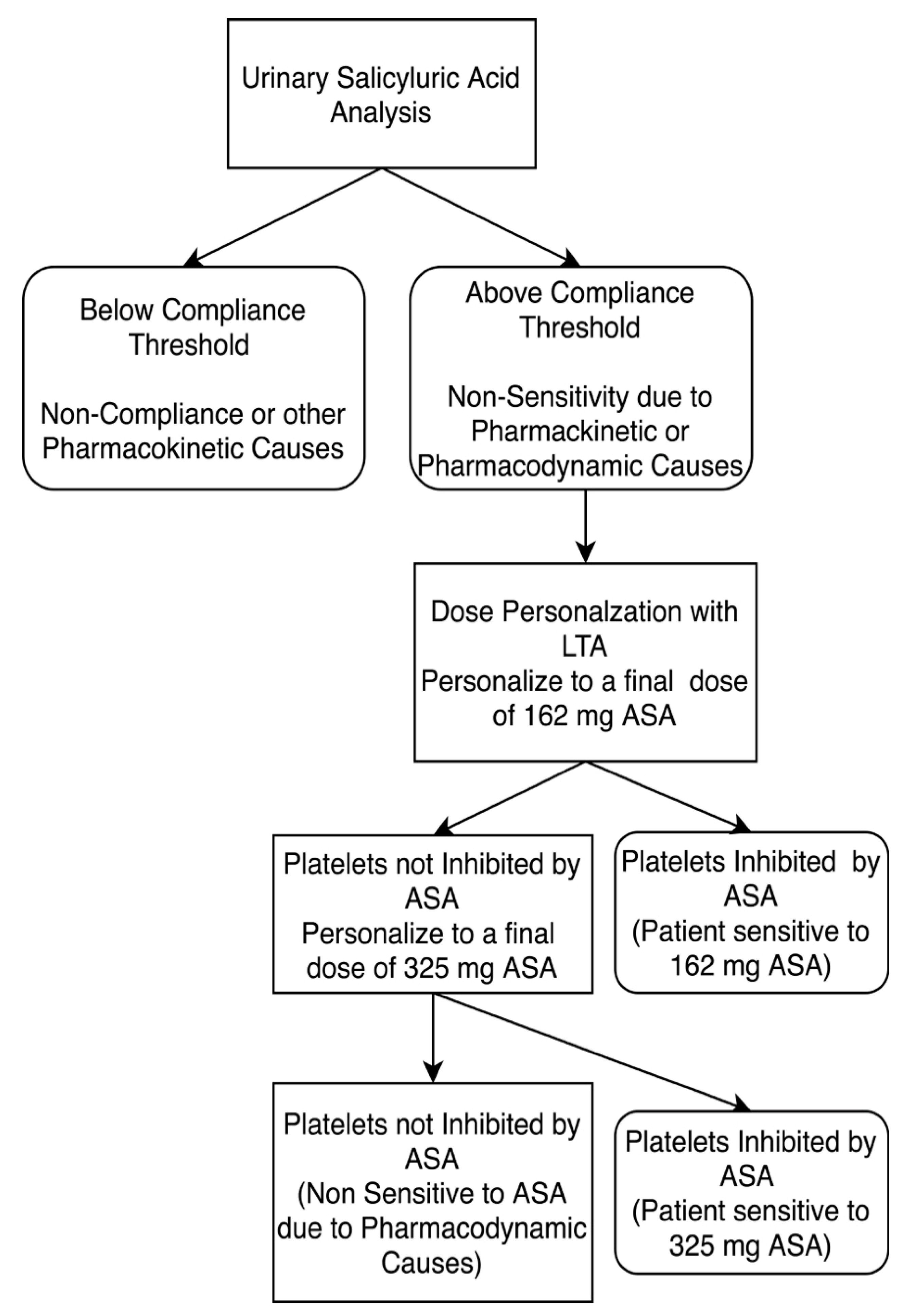
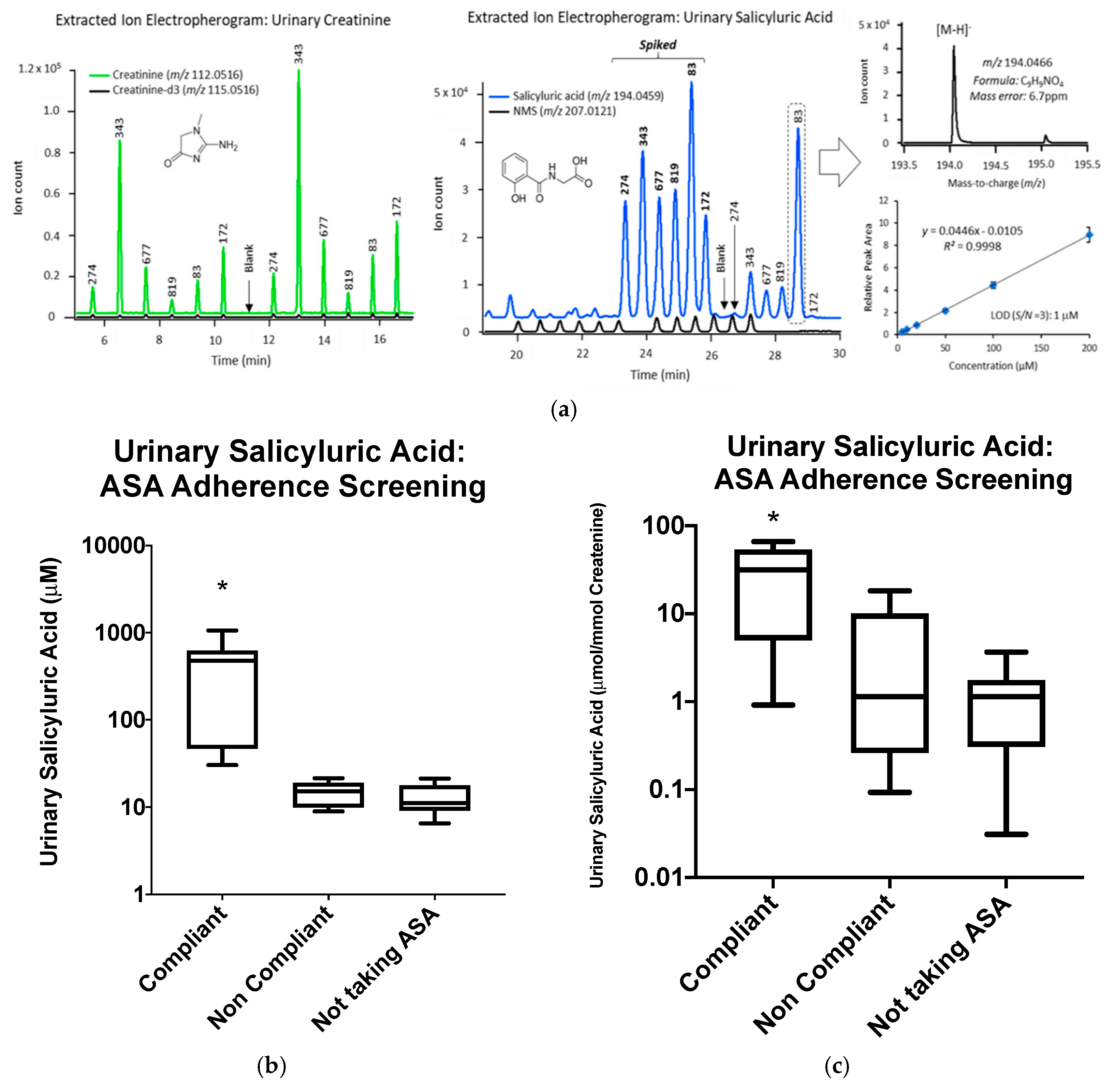
2.5. Urine Salicyluric Acid Analysis
2.6. Ex Vivo Aspirin Personalization
2.7. Statistical Analysis
3. Results
3.1. Cohort Description
3.2. Baseline ASA Sensitivity Testing
3.3. Investigating Possible Mechanisms for Lack of Platelet Inhibition by ASA
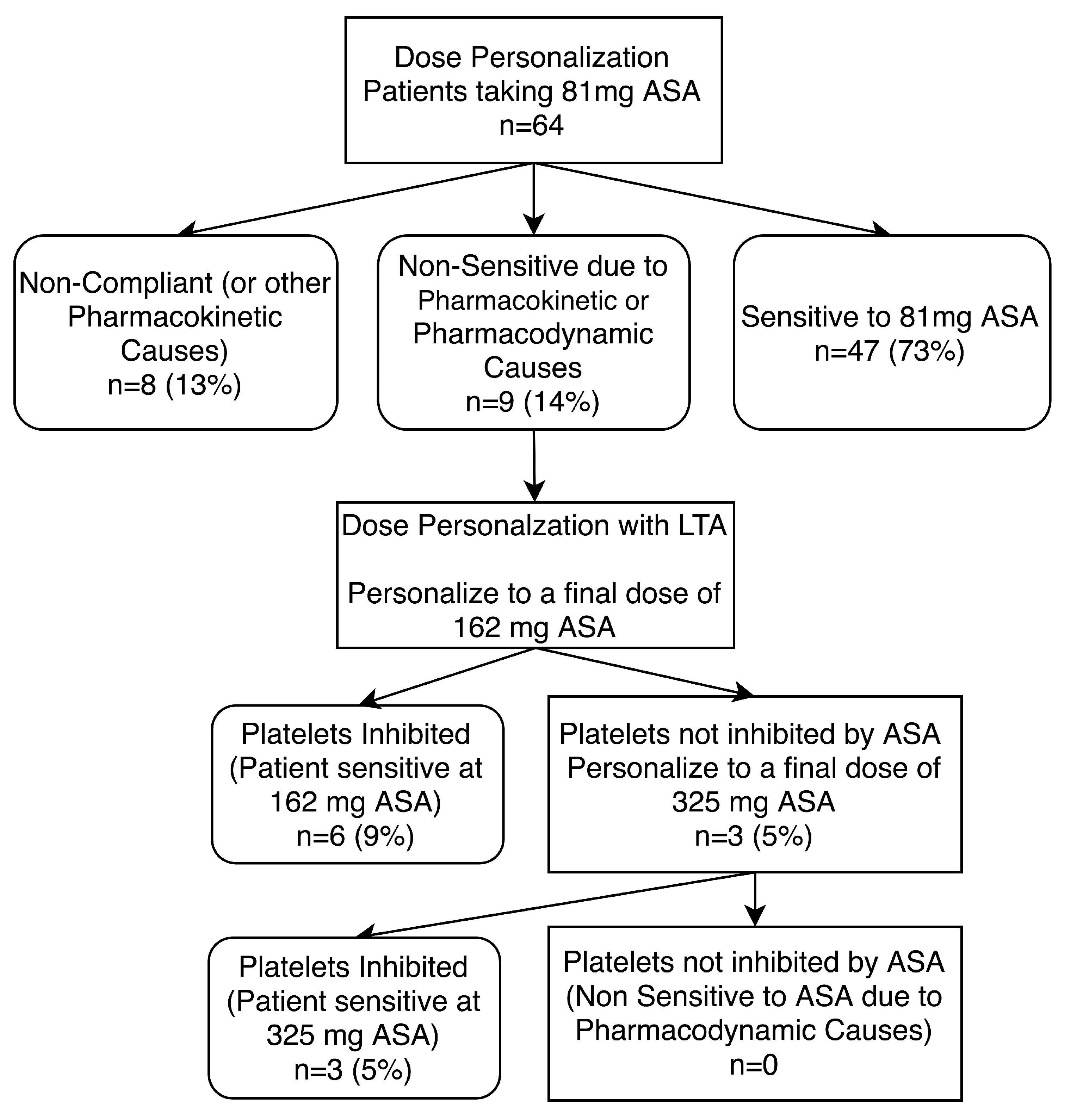
3.4. ASA Dosage Personalization
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Methodology
Appendix A.2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Atherosclerotic Patients on Not Taking ASA (n = 12) | Non-Atherosclerotic Patients on 81 mg ASA (n = 14) |
|---|---|---|
| Mean (SD) | ||
| ABI | 0.63 ± 1.9 | 1.07 ± 0.2 |
| Age (yrs) | 73 ± 11 | 65 ± 17 |
| Platelet Count (103/μL) | 304 ± 79 | 231 ± 56 |
| Frequency (%) | ||
| Sex (% Male) | 83 | 86 |
| Hypertension (%) | 83 | 43 |
| Hyperlipidemia (%) | 83 | 57 |
| Diabetes (%) | 25 | 14 |
| Renal Insufficiency (%) | 8 | 7 |
| Smokers (%) | 83 | 71 |
| CAD (%) | 8 | 0 |
| Patients taking a Statin (%) | 67 | 50 |
| Patients taking an ACEi/ARB (%) | 58 | 28 |
| Patients taking a BB (%) | 8 | 29 |
| Patients taking a CCB (%) | 17 | 14 |
Appendix A.3. Urinary Salicyluric Acid Analysis

References
- Xu, X.R.; Zhang, D.; Oswald, B.E.; Carrim, N.; Wang, X.; Hou, Y.; Zhang, Q.; LaValle, C.; McKeown, T.; Marshall, A.H.; et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit. Rev. Clin. Lab. Sci. 2016, 53, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Carrim, N.; Li, B.X.; Ni, H.; Neves, M.A.D.; McKeown, T.; Stratton, T.W.; Coelho, R.M.P.; Lei, X.; Chen, P.; et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 2016, 14 (Suppl. S1), 29. [Google Scholar] [CrossRef] [PubMed]
- Ricotta, J.J.; Aburahma, A.; Ascher, E.; Eskandari, M.; Faries, P.; Lal, B.K. Updated Society for Vascular Surgery guidelines for management of extracranial carotid disease: Executive summary. J. Vasc. Surg. 2011, 54, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.S.; Pomposelli, F.B.; Clair, D.G.; Geraghty, P.J.; McKinsey, J.F.; Mills, J.L.; Moneta, G.L.; Murad, M.H.; Powell, R.J.; Reed, A.B.; et al. Society for Vascular Surgery practice guidelines for atherosclerotic occlusive disease of the lower extremities: Management of asymptomatic disease and claudication. J. Vasc. Surg. 2015, 61, 2S–41S.e1. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, 596–646. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Chuchalin, A.G.; Eisebitt, R.; Netayzhenko, V.Z.; Voelker, M. Aspirin Compared with Acetaminophen in the Treatment of Feverand OtherSymptoms of Upper Respiratory Tract Infection in Adults: A Multicenter, Randomized, Double-Blind, Double-Dummy, Placebo-Controlled, Parallel-Group, Single-Dose, 6-Hour Dose-Ranging Study. Clin. Ther. 2005, 27, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef]
- Schwartz, K.A. Aspirin Resistance. Neurohospitalist 2011, 1, 94–103. [Google Scholar] [CrossRef]
- Clavijo, L.C.; Al-Asady, N.; Dhillon, A.S.; Matthews, R.V.; Caro, J.; Tun, H.; Rowe, V.L.; Shavelle, D.M. Prevalence of high on-treatment (aspirin and clopidogrel) platelet reactivity in patients with critical limb ischemia. Cardiovasc. Revascularization Med. 2018, 19, 516–520. [Google Scholar] [CrossRef]
- Guirgis, M.; Thompson, P.; Jansen, S. Review of aspirin and clopidogrel resistance in peripheral arterial disease. J. Vasc. Surg. 2017, 66, 1576–1586. [Google Scholar] [CrossRef]
- Pasala, T.; Hoo, J.S.; Lockhart, M.K.; Waheed, R.; Sengodan, P.; Alexander, J.; Gandhi, S. Aspirin Resistance Predicts Adverse Cardiovascular Events in Patients with Symptomatic Peripheral Artery Disease. Tex. Heart Inst. J. 2016, 43, 482–487. [Google Scholar] [CrossRef]
- Krasopoulos, G.; Brister, S.J.; Beattie, W.S.; Buchanan, M.R. Aspirin “resistance” and risk of cardiovascular morbidity: Systematic review and meta-analysis. BMJ 2008, 336, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Westphal, E.S.; Rainka, M.; Amsler, M.; Aladeen, T.; Wisniewski, C.; Bates, V.; Gengo, F.M. Prospective Determination of Aspirin Sensitivity in Patients Resistant to Low Dose Aspirin: A Proof of Concept Study. J. Clin. Pharmacol. 2018, 58, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Petrucci, G. Variability in the Responsiveness to Low-Dose Aspirin: Pharmacological and Disease-Related Mechanisms. Thrombosis 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Paniccia, R.; Antonucci, E.; Gori, A.M.; Marcucci, R.; Poli, S.; Romano, E.; Valente, S.; Giglioli, C.; Fedi, S.; Gensini, G.F.; et al. Comparison of Different Methods to Evaluate the Effect of Aspirin on Platelet Function in High-Risk Patients With Ischemic Heart Disease Receiving Dual Antiplatelet Treatment. Am. J. Clin. Pathol. 2007, 128, 143–149. [Google Scholar] [CrossRef]
- Cattaneo, M. High on-treatment platelet reactivity—Definition and measurement. Thromb. Haemost. 2013, 109, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.; Quinn, T.; Rafferty, M.; Higgins, P.; Ray, G.; Lees, K.R.; Walters, M.R. Aspirin Resistance and Compliance with Therapy. Cardiovasc. Ther. 2011, 29, 301–307. [Google Scholar] [CrossRef]
- Winter, M.-P.; Koziński, M.; Kubica, J.; Aradi, D.; Siller-Matula, J.M. Personalized antiplatelet therapy with P2Y12 receptor inhibitors: Benefits and pitfalls. Postępy Kardiol. Interwencyjnej Adv. Interv. Cardiol. 2015, 11, 259–280. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Wu, T.-T.; Yang, Y.-N.; Hou, X.-G.; Gao, Y.; Chen, Y.; Li, X.-M.; Ma, X.; Ma, Y.-T.; Xie, X. Personalized antiplatelet therapy guided by a novel detection of platelet aggregation function in stable coronary artery disease patients undergoing percutaneous coronary intervention: A randomized controlled clinical trial. Eur. Hear. J. Cardiovasc. Pharmacother. 2020, 6, 211–221. [Google Scholar] [CrossRef]
- Valenti, R.; Marcucci, R.; Comito, V.; Marrani, M.; Cantini, G.; Migliorini, A.; Parodi, G.; Gensini, G.F.; Abbate, R.; Antoniucci, D. Prasugrel in Clopidogrel Nonresponders Undergoing Percutaneous Coronary Intervention. JACC Cardiovasc. Interv. 2015, 8, 1563–1570. [Google Scholar] [CrossRef]
- Xie, X.; Ma, Y.-T.; Yang, Y.-N.; Li, X.-M.; Zheng, Y.-Y.; Ma, X.; Fu, Z.-Y.; Bayinsilema, B.; Li, Y.; Yu, Z.-X.; et al. Personalized antiplatelet therapy according to CYP2C19 genotype after percutaneous coronary intervention: A randomized control trial. Int. J. Cardiol. 2013, 168, 3736–3740. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Tantry, U.S.; Shuldiner, A.R.; Kereiakes, D.J. Genotyping: One Piece of the Puzzle to Personalize Antiplatelet Therapy. J. Am. Coll. Cardiol. 2010, 56, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.H.; Zamzam, A.; Valencia, J.; Khan, H.; Jain, S.; Singh, K.K.; Abdin, R.; Qadura, M. MicroRNA Profile of Patients with Chronic Limb-Threatening Ischemia. Diagnostics 2020, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Wang, Y.; Adili, R.; Ju, L.; Spring, C.M.; Jin, J.W.; Yang, H.; Neves, M.A.D.; Chen, P.; Yang, Y.; et al. Apolipoprotein A-IV binds αIIbβ3 integrin and inhibits thrombosis. Nat. Commun. 2018, 9, 3608. [Google Scholar] [CrossRef]
- Pedersen, S.B.; Grove, E.L.; Nielsen, H.L.; Mortensen, J.; Kristensen, S.D.; Hvas, A.-M. Evaluation of aspirin response by Multiplate® whole blood aggregometry and light transmission aggregometry. Platelets 2009, 20, 415–420. [Google Scholar] [CrossRef]
- Yang, H.; Reheman, A.; Chen, P.; Zhu, G.; Hynes, R.O.; Freedman, J.; Wagner, D.D.; Ni, H. Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo. J. Thromb. Haemost. 2006, 4, 2230–2237. [Google Scholar] [CrossRef]
- Wang, Y.; Reheman, A.; Spring, C.M.; Kalantari, J.; Marshall, A.H.; Wolberg, A.S.; Gross, P.L.; Weitz, J.I.; Rand, M.L.; Mosher, D.F.; et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J. Clin. Investig. 2014, 124, 4281–4293. [Google Scholar] [CrossRef]
- Lordkipanidzé, M.; Pharand, C.; Schampaert, E.; Turgeon, J.; Palisaitis, D.A.; Diodati, J.G. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur. Heart J. 2007, 28, 1702–1708. [Google Scholar] [CrossRef]
- Gum, P.A.; Kottke-Marchant, K.; Poggio, E.D.; Gurm, H.; Welsh, P.A.; Brooks, L.; Sapp, S.K.; Topol, E.J. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am. J. Cardiol. 2001, 88, 230–235. [Google Scholar] [CrossRef]
- Harrison, P.; Segal, H.; Blasbery, K.; Furtado, C.; Silver, L.; Rothwell, P.M. Screening for Aspirin Responsiveness After Transient Ischemic Attack and Stroke. Stroke 2005, 36, 1001–1005. [Google Scholar] [CrossRef]
- Maree, A.O.; Curtin, R.J.; Dooley, M.; Conroy, R.M.; Crean, P.; Cox, D.; Fitzgerald, D.J. Platelet Response to Low-Dose Enteric-Coated Aspirin in Patients With Stable Cardiovascular Disease. J. Am. Coll. Cardiol. 2005, 46, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Tantry, U.S.; Bliden, K.P.; Gurbel, P.A. Overestimation of Platelet Aspirin Resistance Detection by Thrombelastograph Platelet Mapping and Validation by Conventional Aggregometry Using Arachidonic Acid Stimulation. J. Am. Coll. Cardiol. 2005, 46, 1705–1709. [Google Scholar] [CrossRef]
- Lev, E.I.; Patel, R.T.; Maresh, K.J.; Guthikonda, S.; Granada, J.; DeLao, T.; Bray, P.F.; Kleiman, N.S. Aspirin and Clopidogrel Drug Response in Patients Undergoing Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2006, 47, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lecka, J.; Rana, M.S.; Sévigny, J. Inhibition of vascular ectonucleotidase activities by the pro-drugs ticlopidine and clopidogrel favours platelet aggregation. Br. J. Pharmacol. 2010, 161, 1150–1160. [Google Scholar] [CrossRef]
- Trenk, D.; Hochholzer, W.; Fromm, M.F.; Chialda, L.-E.; Pahl, A.; Valina, C.M.; Stratz, C.; Schmiebusch, P.; Bestehorn, H.-P.; Büttner, H.J.; et al. Cytochrome P450 2C19 681G>A Polymorphism and High On-Clopidogrel Platelet Reactivity Associated With Adverse 1-Year Clinical Outcome of Elective Percutaneous Coronary Intervention With Drug-Eluting or Bare-Metal Stents. J. Am. Coll. Cardiol. 2008, 51, 1925–1934. [Google Scholar] [CrossRef]
- Saoi, M.; Li, A.; McGlory, C.; Stokes, T.; Von Allmen, M.T.; Phillips, S.M.; Britz-McKibbin, P. Metabolic Perturbations from Step Reduction in Older Persons at Risk for Sarcopenia: Plasma Biomarkers of Abrupt Changes in Physical Activity. Metabolites 2019, 9, 134. [Google Scholar] [CrossRef]
- Yamamoto, M.; Pinto-Sanchez, M.I.; Bercik, P.; Britz-McKibbin, P. Metabolomics reveals elevated urinary excretion of collagen degradation and epithelial cell turnover products in irritable bowel syndrome patients. Metabolomics 2019, 15, 82. [Google Scholar] [CrossRef]
- Baxter, G.; Lawrence, J.; Graham, A.; Wiles, D.; Paterson, J. Identification and determination of salicylic acid and salicyluric acid in urine of people not taking salicylate drugs. Ann. Clin. Biochem. 2002, 39, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.R.; Peter, R.; Baxter, G.J.; Robson, J.; Graham, A.B.; Paterson, J.R. Urinary excretion of salicyluric and salicylic acids by non-vegetarians, vegetarians, and patients taking low dose aspirin. J. Clin. Pathol. 2003, 56, 651–653. [Google Scholar] [CrossRef]
- Rosenkranz, B.; Frölich, J. Plasma concentrations and anti-platelet effects after low dose acetylsalicylic acid. Prostaglandins Leukot. Med. 1985, 19, 289–300. [Google Scholar] [CrossRef]
- Blacklock, C.J.; Lawrence, J.R.; Wiles, D.; Malcolm, E.A.; Gibson, I.H.; Kelly, C.J.; Paterson, J.R. Salicylic acid in the serum of subjects not taking aspirin. Comparison of salicylic acid concentrations in the serum of vegetarians, non-vegetarians, and patients taking low dose aspirin. J. Clin. Pathol. 2001, 54, 553–555. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Aspirin resistance. Lancet 2006, 367, 606–617. [Google Scholar] [CrossRef]
- Guthikonda, S.; Lev, E.I.; Patel, R.; Delao, T.; Bergeron, A.L.; Dong, J.-F.; Kleiman, N.S. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J. Thromb. Haemost. 2007, 5, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Floyd, C.N.; Ferro, A. Mechanisms of aspirin resistance. Pharmacol. Ther. 2014, 141, 69–78. [Google Scholar] [CrossRef]
- DiChiara, J.; Bliden, K.P.; Tantry, U.S.; Hamed, M.S.; Antonino, M.J.; Suarez, T.A.; Bailon, O.; Singla, A.; Gurbel, P.A. The Effect of Aspirin Dosing on Platelet Function in Diabetic and Nondiabetic Patients: An Analysis From the Aspirin-Induced Platelet Effect (ASPECT) Study. Diabetes 2007, 56, 3014–3019. [Google Scholar] [CrossRef]
- Farrell, B.; Godwin, J.; Richards, S.; Warlow, C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: Final results. J. Neurol. Neurosurg. Psychiatry 1991, 54, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Uk-Tia Study Group. United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: Interim results. Br. Med. J. Clin. Res. Ed. 1988, 296, 316–320. [Google Scholar] [CrossRef]
- Antiplatelet Trialists’ Collaboration. Collaborative overview of randomised trials of antiplatelet therapy--I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ 1994, 308, 81–106. [Google Scholar] [CrossRef]
- Hutt, A.J.; Caldwell, J.; Smith, R.L. The metabolism of aspirin in man: A population study. Xenobiotica 1986, 16, 239–249. [Google Scholar] [CrossRef]
- Yeomans, N.; Lanas, A.; Labenz, J.; van Zanten, S.V.; van Rensburg, C.; Rácz, I.; Tchernev, K.; Karamanolis, D.; Roda, E.; Hawkey, C.; et al. Efficacy of Esomeprazole (20 mg Once Daily) for Reducing the Risk of Gastroduodenal Ulcers Associated With Continuous Use of Low-Dose Aspirin. Am. J. Gastroenterol. 2008, 103, 2465–2473. [Google Scholar] [CrossRef]
- Cohen, H.W.; Crandall, J.P.; Hailpern, S.M.; Billett, H.H. Aspirin resistance associated with HbA1c and obesity in diabetic patients. J. Diabetes Complicat. 2008, 22, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Herzog, W.; Drolet, M.; Pakyz, R.; Newcomer, S.; Sack, P.; Karon, H.; Ryan, K.A.; Zhao, Y.; Shi, X.; et al. Aspirin Resistance in Healthy Drug-Naive Men Versus Women (from the Heredity and Phenotype Intervention Heart Study). Am. J. Cardiol. 2009, 104, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hao, W.-J.; Gao, L.-G.; Chen, T.-M.; Liu, L.; Sun, Y.-F.; Hu, G.-L.; Hu, Y.-X.; Fan, L. Establishing a predictive model for aspirin resistance in elderly Chinese patients with chronic cardiovascular disease. J. Geriatr. Cardiol. JGC 2016, 13, 458–464. [Google Scholar] [PubMed]
- Lee, P.-Y.; Chen, W.-H.; Ng, W.; Cheng, X.; Kwok, J.Y.-Y.; Tse, H.-F.; Lau, C.-P. Low-dose aspirin increases aspirin resistance in patients with coronary artery disease. Am. J. Med. 2005, 118, 723–727. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]







| Characteristics | Atherosclerotic Patients on 81 mg ASA (n = 64) |
|---|---|
| Mean (SD) | |
| ABI | 0.6 ± 0.19 |
| Age (yrs) | 71 ± 8 |
| Platelet Count (103/μL) | 252 ± 62 |
| Frequency (%) | |
| Sex (% Male) | 54 |
| Hypertension (%) | 71 |
| Hyperlipidemia (%) | 88 |
| Diabetes (%) | 45 |
| Renal Insufficiency (%) | 7 |
| Smokers (%) | 88 |
| CAD (%) | 32 |
| Patients taking a Statin (%) | 73 |
| Patients taking an ACEi/ARB (%) | 50 |
| Patients taking a BB (%) | 27 |
| Patients taking a CCB (%) | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, H.; Gallant, R.C.; Zamzam, A.; Jain, S.; Afxentiou, S.; Syed, M.; Kroezen, Z.; Shanmuganathan, M.; Britz-McKibbin, P.; Rand, M.L.; et al. Personalization of Aspirin Therapy Ex Vivo in Patients with Atherosclerosis Using Light Transmission Aggregometry. Diagnostics 2020, 10, 871. https://doi.org/10.3390/diagnostics10110871
Khan H, Gallant RC, Zamzam A, Jain S, Afxentiou S, Syed M, Kroezen Z, Shanmuganathan M, Britz-McKibbin P, Rand ML, et al. Personalization of Aspirin Therapy Ex Vivo in Patients with Atherosclerosis Using Light Transmission Aggregometry. Diagnostics. 2020; 10(11):871. https://doi.org/10.3390/diagnostics10110871
Chicago/Turabian StyleKhan, Hamzah, Reid C. Gallant, Abdelrahman Zamzam, Shubha Jain, Sherri Afxentiou, Muzammil Syed, Zachary Kroezen, Meera Shanmuganathan, Philip Britz-McKibbin, Margaret L. Rand, and et al. 2020. "Personalization of Aspirin Therapy Ex Vivo in Patients with Atherosclerosis Using Light Transmission Aggregometry" Diagnostics 10, no. 11: 871. https://doi.org/10.3390/diagnostics10110871
APA StyleKhan, H., Gallant, R. C., Zamzam, A., Jain, S., Afxentiou, S., Syed, M., Kroezen, Z., Shanmuganathan, M., Britz-McKibbin, P., Rand, M. L., Ni, H., Al-Omran, M., & Qadura, M. (2020). Personalization of Aspirin Therapy Ex Vivo in Patients with Atherosclerosis Using Light Transmission Aggregometry. Diagnostics, 10(11), 871. https://doi.org/10.3390/diagnostics10110871