1. Introduction
Molybdenum cofactor deficiency type B (MOCODB, #252160) is an autosomal recessive metabolic disorder characterized by intractable seizures of neonatal-onset, muscular spasticity, accompanied by hypouricemia, elevated urinary sulfite levels and craniofacial dysmorphism. It first came to medical attention in 1980 [
1]. Affected children show severe neurologic complications, which may lead to early death, and rarely (only seven cases described to date) present with a milder form with global developmental delay without seizures [
2,
3]. The severe manifestation of the disease is characterized by neonatal-onset, intractable seizures, severe feeding difficulties, accompany by progressive neurological deterioration (opisthotonos, hypertonicity, spastic quadriplegia) and cystic leukomalacia in brain MR. The milder phenotype is defined with longer surveillance and first occurrence of seizures after the neonatal period. In both types, dysmorphic facial features may be noted, as a long face with frontal bossing, hypertelorism, puffy cheeks and long philtrum. The disorder results from decreased activity of sulfite oxidase (SUOX; EC 1.8.3.1) and xanthine dehydrogenase (XDH; EC 1.17.1.4 and 1.17.3.2), which are molybdenum cofactor-dependent for their activity, while molybdenum cofactor (MoCo) is essential also for aldehyde oxidase activity (AOX1; EC 1.2.3.1). It is mainly sulfite oxidase deficiency that will lead to the severe clinical presentation.
Molybdenum cofactor (MOCS2) is encoded by the
MOCS2 gene, localized on chromosome 5q11.2. Its biosynthesis is, however, a multistep process, involving the synthesis of precursor Z by proteins encoded by
MOCS1 (603707), conversion of precursor Z to molybdopterin (MPT) by MPT synthase (MOCS2), and attachment of molybdenum to the dithiolene moiety of MPT by gephyrin (GPHN; EC 2.10.1.1 and 2.7.7.75) [
4,
5]. MOCS2 is bicistronic, overlapping open reading frames (ORFs) which encode MOCS2A and MOCS2B, the two subunits of MPT synthase [
6], which implicates its functioning and molecular diagnostics of MOCS2 deficiency.
In this paper, we seek present another patient, in whom a novel, in-frame variant within the MOCS2 gene was identified. We aim to delineate the MOCS2 phenotype and give evidence of the clinical significance of the novel variant.
2. Clinical Description and Methods
The patient was born from second pregnancy by Caesarean section (due to polyhydramnios, fetal heart rate anomalies in cardiotocography) in the 38th week with a birth weight of 3960 g, and head circumference of 37 cm (>97th percentile). In the Apgar scale, he received 7/7/10/10 points. Irregular breathing, pale grey skin, and increased muscle tone were noted; the saturation was 65% and there was mild metabolic acidosis in umbilical cord blood. Mechanical ventilation was used to stabilize the child’s condition. During pregnancy, the mother was diagnosed with hypothyroidism, treated with levothyroxine; fetal hiccups were noted. The family history for miscarriages, deaths in early childhood, and neurological and metabolic diseases were negative. The child’s parents are unrelated; the proband has a healthy, 2-year-old sister.
Physical examination revealed facial dysmorphism (such as coarse features with puffy cheeks, bitemporal narrowing, deep-set eyes, long philtrum, dysplastic auricles), long and overlapping fingers, large feet, pectus excavatum, swelling of the eyelids and lumbosacral region. At 30 min after the birth, deterioration in the general condition and grunting drops in desaturation were observed with subsequent diagnosis of congenital pneumonia. Difficulties in feeding (lack of sucking), muscular tension disorders, epileptic seizures, and breathing difficulties with an abnormal crying further complicated the perinatal period. Convulsions with drops in saturation occurred on the first day of life. Despite the phenobarbital implementation, polymorphic epileptic seizures, limb myoclonus, breathing disorders, sometimes with desaturation, were still observed. Within the following days, the child became inactive and flaccid. Abnormal spontaneous movements of the extremities were observed, such as nonrhythmic waving, pedaling, and increased muscle tone, especially in upper extremities. Reduced muscular tension was accompanied by areflexia. Since seizures were still uncontrolled, levetiracetam was additionally introduced to the treatment with initially good effect. During the hospitalization, muscle tone gradually increased, and elbow and knee joints contractures appeared.
In the second month of life, the child gradually became more reactive, but still presented clearly an impoverished spontaneous motor activity. At times, the boy opened his eyes, but with no eye contact and did not follow with his eyes. He was fed with a nasogastric tube. Physically, the skin was pale, pasty, head circumference was still above the 97th percentile, and prominent cranial sutures were noted. The axial hypotonia and lower limb spasticity were observed; tendon hyperreflexia appeared. In the neurological examination, the traction test was negative, no sole reflex on both sides, no Babinski sign bilaterally, weak grasping reflexes, no Moro reflex. Periodical restlessness, crying, and hyperekplexia were observed, as well as epileptic seizures, mainly myoclonic. Thus, in addition to phenobarbital and levetiracetam, valproic acid was later introduced. Due to tachycardia, propranolol was included in the treatment. Hypothyroidism requiring substitution with levothyroxine was also diagnosed. Because of his severe and worsening clinical condition, the child was transferred to a home hospice.
The laboratory and imagine assessment were summarized in
Table 1.
In order to verify the clinical diagnosis—deficiency of molybdenum cofactor—taken in terms of disease manifestation and laboratory result, genetic testing was warranted. However, given the nonspecific features (macrocephaly at birth, hypertrophic cardiomyopathy and lack of cystic leukomalacia in brain imagining), we decided on exome sequencing.
2.1. Methods
2.1.1. Molecular Analysis
Whole exome sequencing (WES) was performed in 3billion, Inc (Seoul, South Korea), using genomic DNA isolated from the patient’s whole blood with informed consent obtained from the patient’s parents; in the light of the applicable law, there was no requirement for ethics committee review. The captured genomic regions (exons of approx. 22,000 genes) were sequenced using a NovaSeq platform (Illumina, San Diego, CA, USA) and aligned to the reference sequence (NCBI genome assembly GRCh37; accessed in February 2009), as previously described [
7]. The mean depth of coverage was 100-fold, with 99.2% coverage higher than 10-fold. Sanger sequencing of the identified variant was performed for the patient.
2.1.2. Crystal Structures Analysis
The crystal structure of human molybdopterin synthase complex (PDB ID: 5MPO [
8]) and molybdopterin synthase from
Staphylococcus aureus complexed with precursor Z (PDB ID: 2QIE [
9]) were downloaded from the Protein Data Bank [
10]. Protein structures were aligned and analyzed using PyMol software (PyMol Version 2.0, Schrödinger LLC, New York, NY, USA). The potential effects of substitutions at 158 and 159 positions in the molybdopterin synthase catalytic subunit were examined by the PredictSNP webserver (
https://loschmidt.chemi.muni.cz/predictsnp/ access date: 11 May 2020) [
11].
3. Results
3.1. Genetic Result
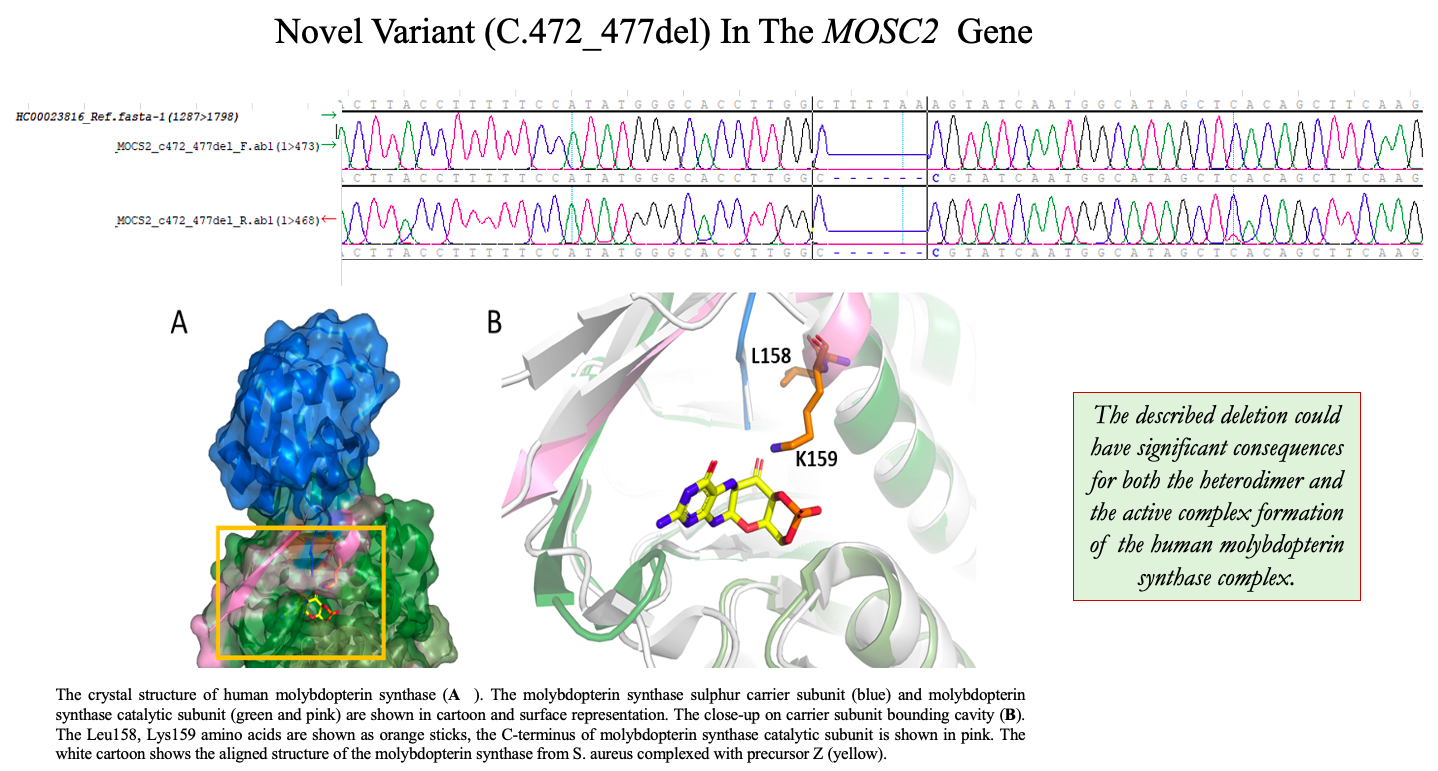
WES led to the identification of a variant in the
MOCS2 gene—c.472_477del (
Table 2,
Figure S1). The results of the predicted effect of mutations at position 158 and 159 of the catalytic subunit of molybdopterin synthase complex obtained by the PredictSNP server [
11] are shown (
Table S1).
3.2. Structural Analysis
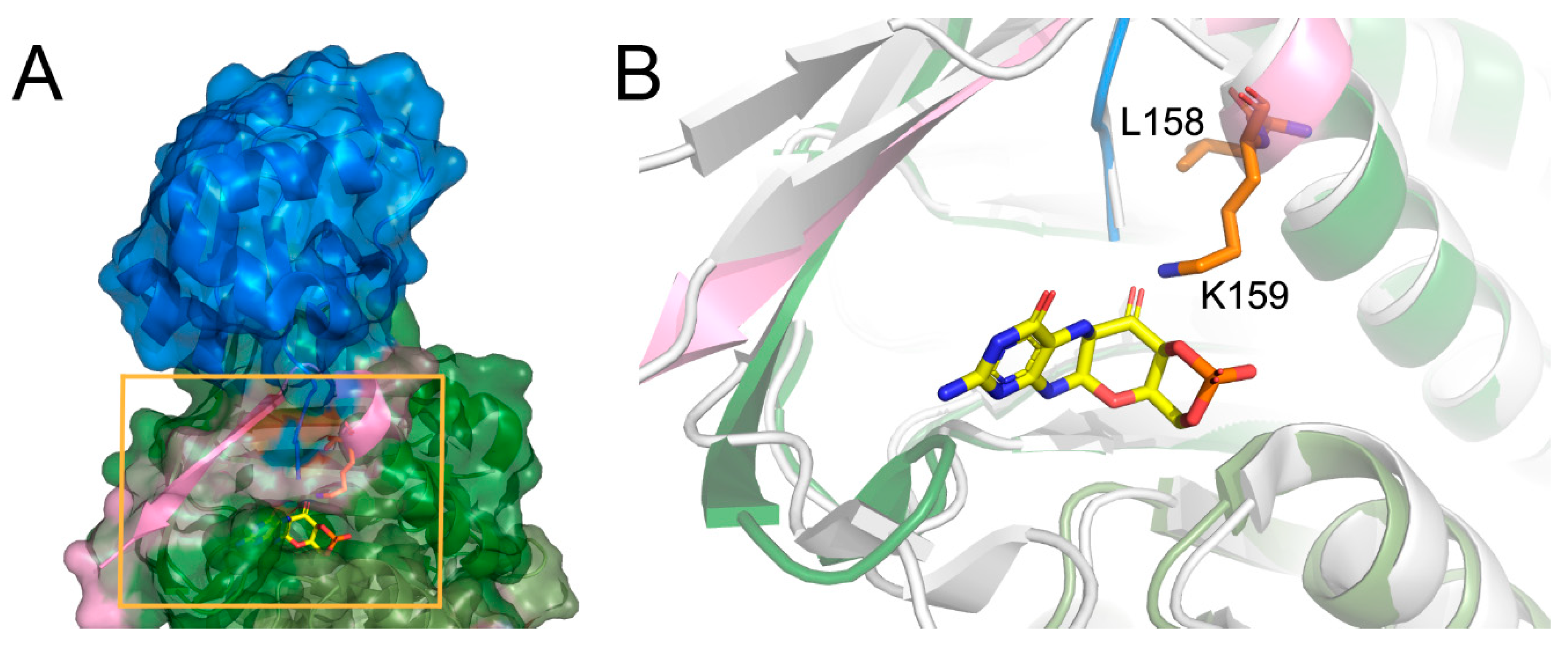
There is no crystal structure of the protein carrying the described deletion; however, the analysis of the native conformation of the molybdopterin synthase and comparison with the structure from
Staphylococcus aureus complexed with precursor Z shed light on the consequences of the deletion. Human molybdopterin synthase complex was crystallized as a dimer constituted of two heterodimers. Each heterodimer consists of molybdopterin synthase sulfur carrier subunit and molybdopterin synthase catalytic subunit. The Leu158 and Lys159 deletion occurs in the catalytic subunit. The Leu158 and Lys159 residues are located at the end of the last helix, close to the C-terminus of the subunit, and together with the C-terminus, they participate in the molybdopterin synthase sulfur carrier subunit binding (
Figure 2A). Moreover, the comparison of the structures of molybdopterin synthases from human and
Staphylococcus aureus suggests that the Lys159 residue is essential for precursor Z binding (
Figure 2B).
4. Discussion
The molybdenum cofactor deficiency is an ultra-rare autosomal recessive disease, characterized by rapidly progressive and severe neurological damage, sometimes misdiagnosed as hypoxic-ischemic encephalopathy. The clinical and molecular characteristics of molybdenum cofactor deficiency due to
the MOCS2 variant have been recently extensively reviewed by Arican et al. [
2]. The presented group encompassed 35 patients with proven molecular etiology and 29 different pathogenic variants identified in the
MOCS2 gene.
Our proband’s history and clinical features, in terms of neonatal seizures and the biochemical test results, are consistent with the previous descriptions. That prompted us to test for a deficiency of molybdenum cofactor, while neonatal-onset, refractory epilepsy, feeding difficulties, and facial dysmorphism were the most suggestive phenotypic features. The proband presented, however, some unique characteristics, not consistent with molybdenum cofactor deficiency, like macrocephaly at birth, hypertrophic cardiomyopathy (restrictive) with elevated troponin and lack of cystic leukomalacia in brain imagining. Under such circumstances, the performance of genetic diagnostics, to a greater extent than targeted testing (of the MOCS2 gene only), was justified. As a result, the initial clinical diagnosis, supported by undetectable serum uric acid, very low serum homocysteine, and positive sulfite dipstick results (the test definitely showed a high concentration of sulfites in the fresh urine; sulfocysteine was not measured, but the purine and pyrimidine profile in urine showed very high xanthine concentration), was finally verified in whole-exome sequencing analysis. It revealed a variant of unknown significance in the MOCS2 gene. The homozygous variant c.472_477del on MOCS2 deletes nucleotides ‘CTTTTA’ from chromosome5 52396264, which does not change frameshift in the protein-coding sequence. It has been reported with an extremely low frequency in the large population cohorts (GenomAD). This in-frame deletion in the non-repeat region can change the length of proteins and disrupt protein function. This variant is classified as of uncertain significance according to the recommendation of the American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP) guideline. The child’s clinical features were, however, highly consistent with the disease. Additionally, taking into account the homozygous state of this variant, we suggest it as being causative. Referring to the additional clinical characteristics, not reported in previous cases, their significance as features of molybdenum cofactor deficiency type B needs to be verified based on additional cases with the same genetic etiology (genotype).
We have performed an analysis of the crystal structure of the human molybdopterin synthase complex to support this statement. The Leu158 and Lys159 are localized at the end of the last helix of the catalytic subunit, just before the 13 aa of the C-terminus. This region of the molybdopterin synthase has dual functionality. It binds the sulfur carrier subunit and the precursor Z. The alignment of the crystal structure of human molybdopterin synthase with the one of Staphylococcus aureus complexed with precursor Z reflects that Lys159 corresponds to Leu116 of (S. aureus) as an essential residue for proper binding of the precursor Z.
As a consequence of the mutation, the Leu158 will be replaced by Ala160, and Lys159 by Lys161. Luckily, the deleted Lys159 should be replaced by Lys161, and thus its role can be preserved (a similar replacement of the key amino acid was observed, for example, in D-amino acid oxidase, where Tyr314 repossesses the function of the Tyr55, eliminated by the Tyr55Ala mutation and modifies the enzyme-substrate specificity) [
12]. Since the second one replaces the crucial lysine, one could expect that the precursor Z binding can still occur; however, the protein–ligand affinity can be already modified. However, in this case, the situation is more complicated. The second lysine residue (123 in
S. aureus and 166 in human) is essential for precursor Z binding, and this crucial residue is located at the C-terminus. It is highly probable that the deletion of two residues can modify the position of the second lysine.
Moreover, it will have consequences on the folding of the C-terminus and can result in changes in binding affinity of the sulfur carrier subunit to the catalytic one. Thus, the described deletion could have significant consequences for both the heterodimer and the active complex formation, which may suggest another mechanism of pathogenicity of the c.472_477del variant in the
MOCS2 gene. As results from predictSNP have shown (
Table S1), no deletion analysis is possible, but the report clearly indicates that any mutation in this area has consequences. However, to verify which functionality is mostly disturbed, the precursor binding or carrier subunit binding, further study has to be considered.
5. Conclusions
In this paper, we give clinical and structural evidence of pathogenicity of the novel, in-frame variant c.472_477del in the MOCS2 gene. We emphasize the need for rapid genetic diagnosis, using next-generation sequencing—rapid WES (whole-exome sequencing)—in metabolic diseases, especially in cases with an unclear clinical picture or when the prognosis is poor. The described mutation modifies the binding cavity of the catalytic subunit, and the preliminary structural analysis shows that two potential processes that can be distorted. We can speculate that if the heterodimer formation is preserved, the deficiency caused by abnormal precursor Z binding could perhaps be limited by such chemical modification of the precursor Z molecule that would restore native complex properties. The second potential mechanism of deficiency, improper carrier subunit binding, would be much more challenging to reverse.

 ,
, 




{kind=link}
{kind=link}
{kind=link}