Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Results
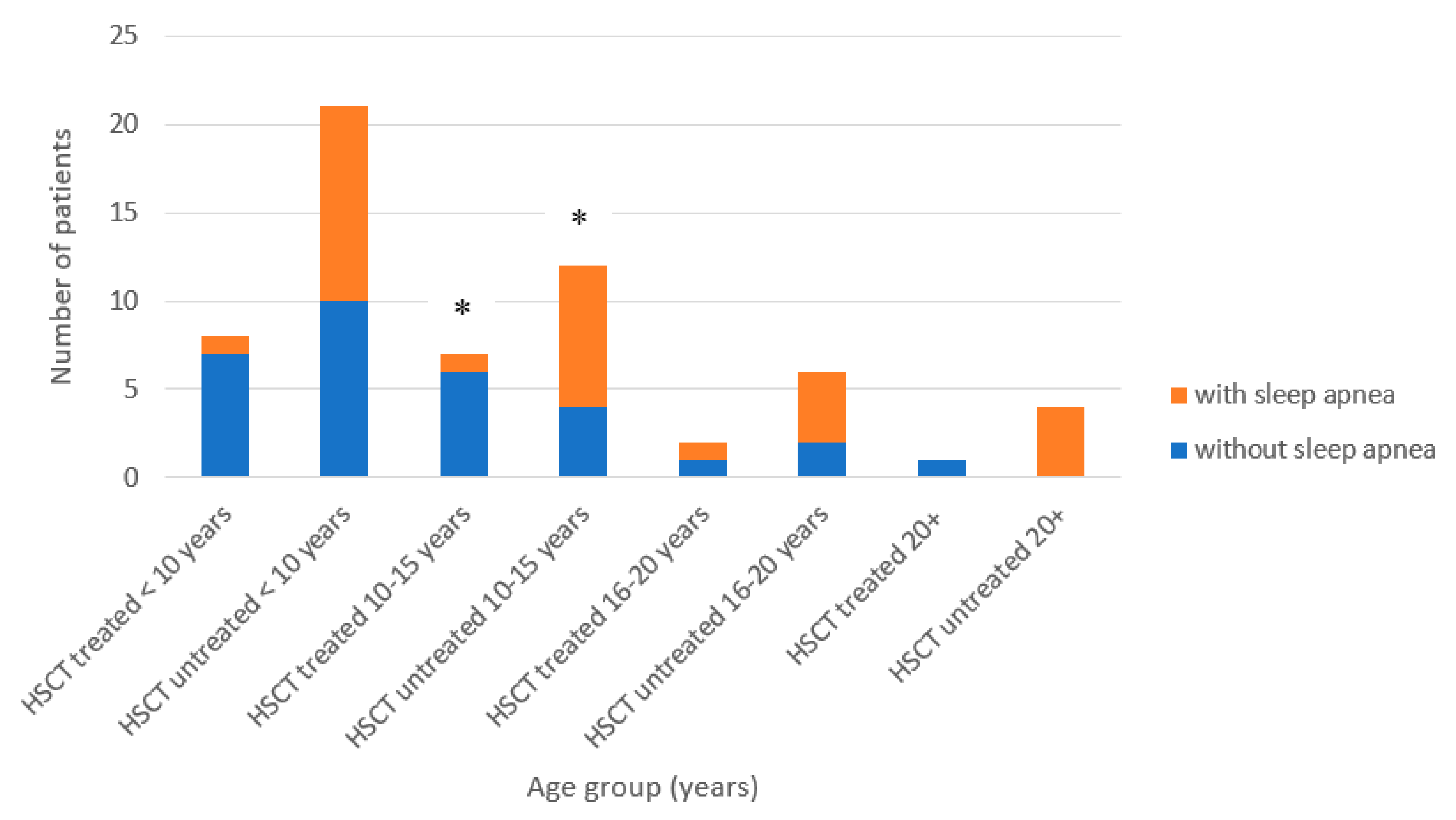
3.1. Patients’ Characteristics and Clinical Manifestations
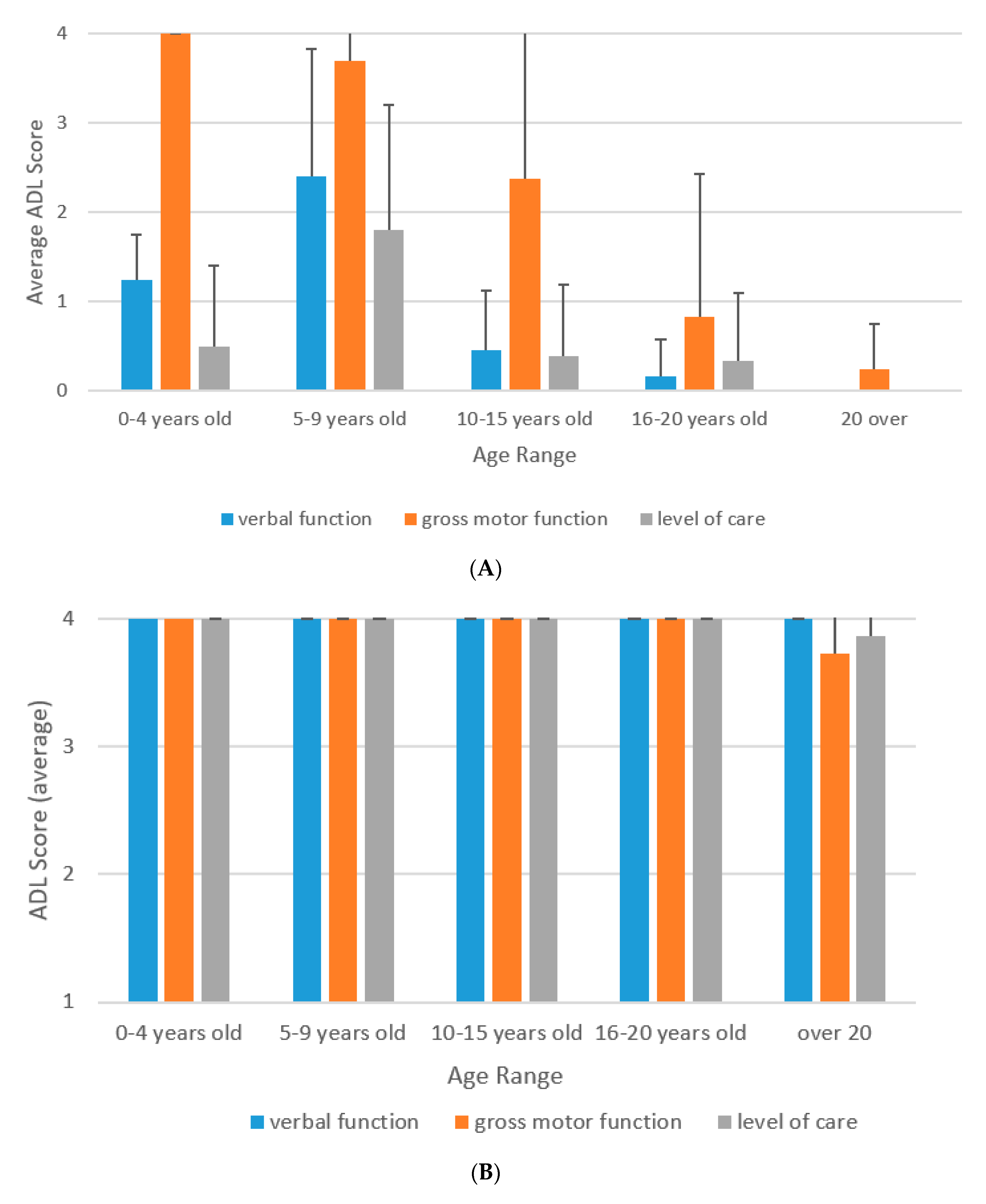
3.2. Natural History of ADL of MPS II
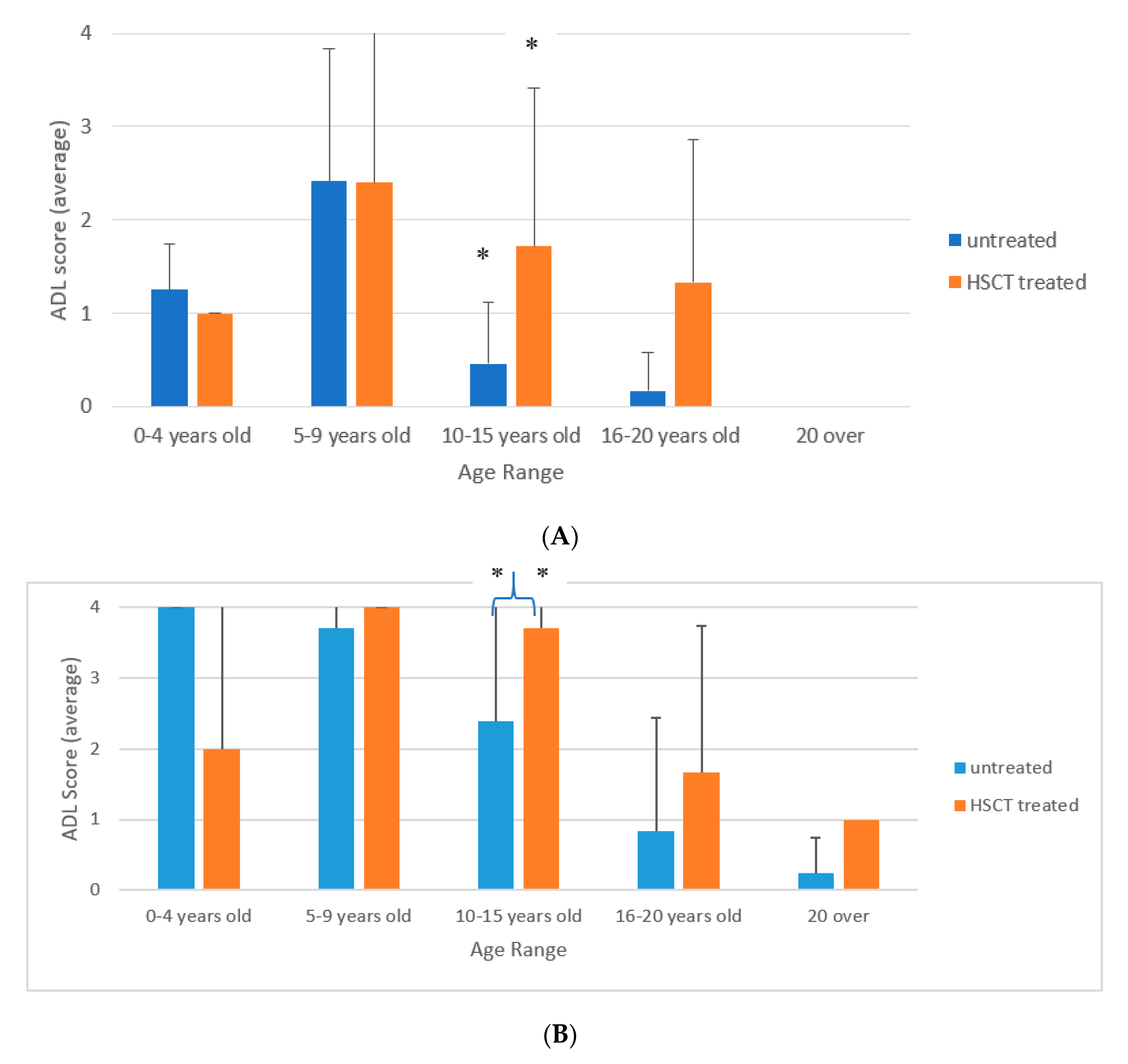
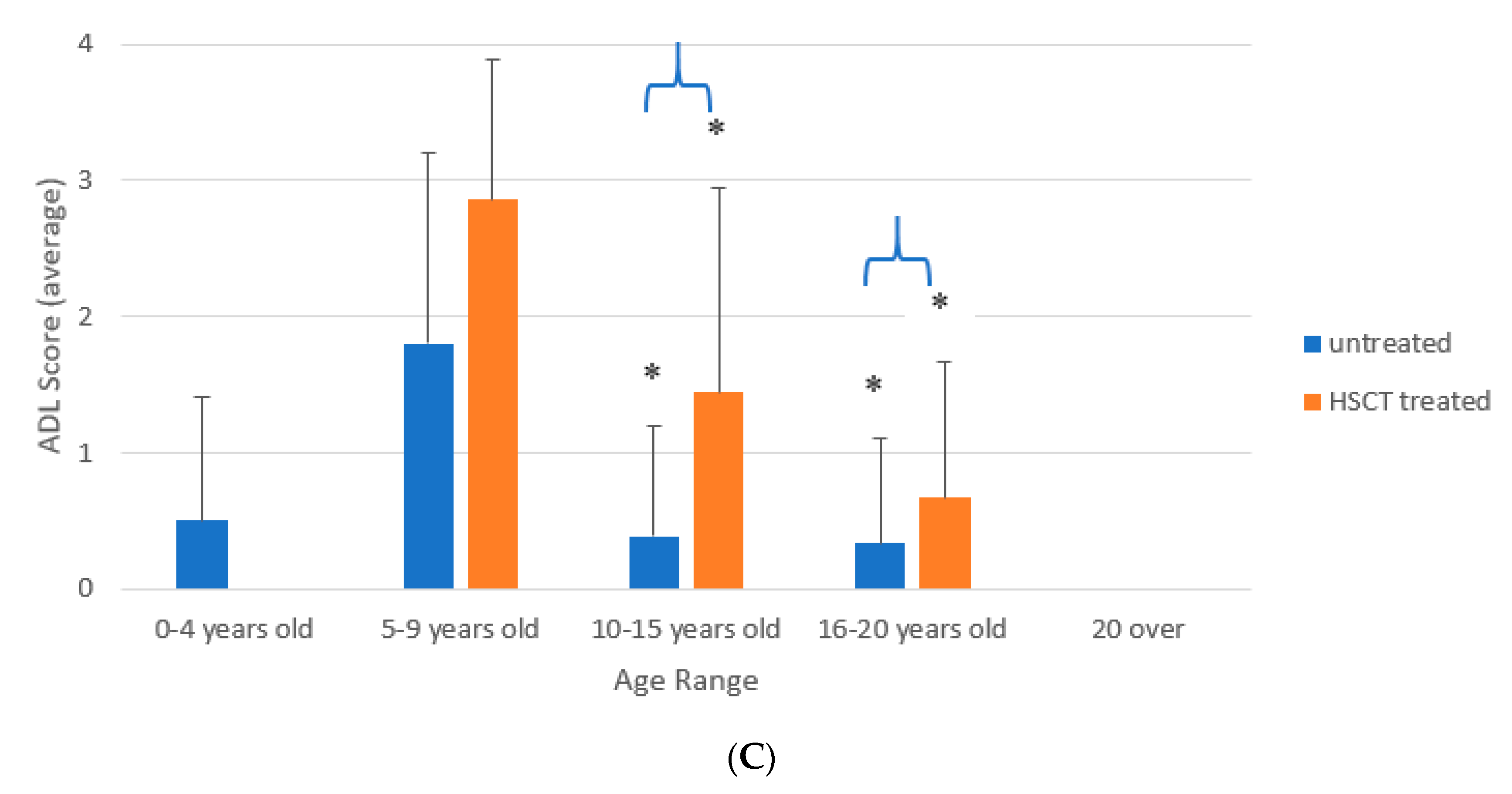
3.3. ADL of Patients Treated with HSCT
4. Discussion
5. Limitations of the Study
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HSCT | Hematopoietic stem cell transplantation |
| MPS II | Mucopolysaccharidosis type II (Hunter disease) |
| ADL | Activity of living |
| GAGs DS HS ERT MRI FIM | Glycosaminoglycans Dermatan sulfate Heparan sulfate Enzyme replacement therapy Magnetic resonance therapy Functional independence measure |
References
- Neufeld, E.; Muenzer, J. The Online Metabolic Basis of Inherited Disease; The McGraw-Hill Metabolic bases of inherited diseases; McGraw-Hill: New York, NY, USA, 2010. [Google Scholar]
- Tomatsu, S.; Kubaski, F.; Stapleton, M.; Suzuki, Y.; Orii, K.O.; Vairo, F.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Graeff Burin, M.; Fischender Moura de Souza, C.; et al. Mucopolysaccharidosis Type II: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment; Nova Science Publishers: New York, NY, USA, 2018; Volume 1, pp. 235–271. [Google Scholar]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Peters, C.; Balthazor, M.; Shapiro, E.G.; King, R.J.; Kollman, C.; Hegland, J.D.; Henslee-Downey, J.; Trigg, M.E.; Cowan, M.J.; Sanders, J.; et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996, 87, 4894–4902. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef] [PubMed]
- McKinnis, E.J.; Sulzbacher, S.; Rutledge, J.C.; Sanders, J.; Scott, C.R. Bone marrow transplantation in Hunter syndrome. J. Pediatr. 1996, 129, 145–148. [Google Scholar] [CrossRef]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef]
- Barth, A.L.; de Magalhaes, T.; Reis, A.B.R.; de Oliveira, M.L.; Scalco, F.B.; Cavalcanti, N.C.; Silva, D.S.E.; Torres, D.A.; Costa, A.A.P.; Bonfim, C.; et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: A 7 years follow-up. Mol. Genet. Metab. Rep. 2017, 12, 62–68. [Google Scholar] [CrossRef]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Kubaski, F.; Stapleton, M.; Suzuki, Y.; Orii, K.O.; Vairo, F.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Burin, M.G.; Souza, C.F.M.D.; et al. Mucopolysaccharidosis type II: Clinical features, biochemistry, diagnosis, genetics, and treatment. In Mucopolysaccharidoses Update; Tomatsu, S., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 165–210. [Google Scholar]
- Tanjuakio, J.; Suzuki, Y.; Patel, P.; Yasuda, E.; Kubaski, F.; Tanaka, A.; Yabe, H.; Mason, R.W.; Montano, A.M.; Orii, K.E.; et al. Activities of daily living in patients with Hunter syndrome: Impact of enzyme replacement therapy and hematopoietic stem cell transplantation. Mol. Genet. Metab. 2015, 114, 161–169. [Google Scholar] [CrossRef]
- Patel, P.; Suzuki, Y.; Maeda, M.; Yasuda, E.; Shimada, T.; Orii, K.E.; Orii, T.; Tomatsu, S. Growth charts for patients with Hunter syndrome. Mol. Genet. Metab. Rep. 2014, 1, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Bodamer, O.; Burton, B.; Clarke, L.; Frenking, G.S.; Giugliani, R.; Jones, S.; Rojas, M.V.; Scarpa, M.; Beck, M.; et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur. J. Pediatr. 2012, 171, 181–188. [Google Scholar] [CrossRef]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 2008, 41, 99–108. [Google Scholar] [CrossRef]
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761103s000lbl.pdf (accessed on 12 December 2019).
- Kato, T.; Kato, Z.; Kuratsubo, I.; Ota, T.; Orii, T.; Kondo, N.; Suzuki, Y. Evaluation of ADL in patients with Hunter disease using FIM score. Brain Dev. 2007, 29, 298–305. [Google Scholar] [CrossRef]
- Peters, C.; Krivit, W. Hematopoietic cell transplantation for mucopolysaccharidosis IIB (Hunter syndrome). Bone Marrow Transplant. 2000, 25, 1097–1099. [Google Scholar] [CrossRef]
- Coppa, G.V.; Gabrielli, O.; Cordiali, R.; Villani, G.R.; Di Natale, P. Bone marrow transplantation in a Hunter patient with P266H mutation. Int. J. Mol. Med. 1999, 4, 433–436. [Google Scholar] [CrossRef]
- Vellodi, A.; Young, E.; Cooper, A.; Lidchi, V.; Winchester, B.; Wraith, J.E. Long-term follow-up following bone marrow transplantation for Hunter disease. J. Inherit. Metab. Dis. 1999, 22, 638–648. [Google Scholar] [CrossRef]
- Guffon, N.; Bertrand, Y.; Forest, I.; Fouilhoux, A.; Froissart, R. Bone marrow transplantation in children with Hunter syndrome: Outcome after 7 to 17 years. J. Pediatr. 2009, 154, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M. Mucopolysaccharidosis Type II. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Boelens, J.J.; van Hasselt, P.M. Neurodevelopmental Outcome after Hematopoietic Cell Transplantation in Inborn Errors of Metabolism: Current Considerations and Future Perspectives. Neuropediatrics 2016, 47, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Tomatsu, S.; Sukegawa, K.; Suzuki, Y.; Kondo, N.; Hopwood, J.J.; Orii, T. Mucopolysaccharidosis type II (Hunter disease): 13 gene mutations in 52 Japanese patients and carrier detection in four families. Hum. Genet. 1993, 92, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Hwang, H.Z.; Song, S.M.; Paik, K.H.; Kwon, E.K.; Moon, K.B.; Yoon, J.H.; Han, C.K.; Jin, D.K. Mutational spectrum of the iduronate 2 sulfatase gene in 25 unrelated Korean Hunter syndrome patients: Identification of 13 novel mutations. Hum. Mutat. 2003, 21, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lin, S.P.; Chuang, C.K.; Niu, D.M.; Chen, M.R.; Tsai, F.J.; Chao, M.C.; Chiu, P.C.; Lin, S.J.; Tsai, L.P.; et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am. J. Med. Genet. A 2009, 149, 960–964. [Google Scholar] [CrossRef]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Khan, S.; Orii, T.; Giugliai, R.; Tomatsu, S. Epidemiology of Mucopolysaccharidoses. In Mucopolysaccharidoses Update; Tomastu, S., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 21–46. [Google Scholar]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef]
- Okuyama, T.; Tanaka, A.; Suzuki, Y.; Ida, H.; Tanaka, T.; Cox, G.F.; Eto, Y.; Orii, T. Japan Elaprase Treatment (JET) study: Idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol. Genet. Metab. 2010, 99, 18–25. [Google Scholar] [CrossRef]
- Mullen, C.A.; Thompson, J.N.; Richard, L.A.; Chan, K.W. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia. Bone Marrow Transplant. 2000, 25, 1093–1097. [Google Scholar] [CrossRef]
- Chang, G.; Ratichek, S.J.; Recklitis, C.; Syrjala, K.; Patel, S.K.; Harris, L.; Rodday, A.M.; Tighiouart, H.; Parsons, S.K. Children’s psychological distress during pediatric HSCT: Parent and child perspectives. Pediatr. Blood Cancer 2012, 58, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Kumar, A.B.; Yi, F.; Liu, Y.; Chennamanen, N.; Masi, S.; Elliot, S.; Turecek, F.; Scott, C.R. Newborn Screening for MPS Diseases by Direct Assay of Enzymes in Dried Blood Spots by Tandem Mass Spectrometry; Nova Science Publishers: New York, NY, USA, 2018; Volume 1, pp. 639–646. [Google Scholar]
- Chuang, C.-K.; Liao, H.-C.; Lin, H.-Y.; Chiang, C.-C.; Lin, S.-P. Newborn Screening of Mucopolysaccharidoses: Comparison of Fluorometric technique and Tandem Mass Spectrometry for Enzyme Assay. In Mucopolysaccharidoses Update; Nova Science Publishers: New York, NY, USA, 2018; Volume 1, pp. 647–660. [Google Scholar]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Newborn screening and biomarkers for Mucopolysaccharidoses by GAG assay. In Mucopolysaccharidoses Update; Nova Science Publishers: New York, NY, USA, 2018; Volume 1, pp. 661–684. [Google Scholar]
- Boado, R.J.; Zhang, Y.; Xia, C.F.; Wang, Y.; Pardridge, W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 99, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Pardridge, W.M. Brain and Organ Uptake in the Rhesus Monkey in Vivo of Recombinant Iduronidase Compared to an Insulin Receptor Antibody-Iduronidase Fusion Protein. Mol. Pharm. 2017, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Boado, R.J.; Giugliani, R.; Schmidt, M. Plasma Pharmacokinetics of Valanafusp Alpha, a Human Insulin Receptor Antibody-Iduronidase Fusion Protein, in Patients with Mucopolysaccharidosis Type I. BioDrugs 2018, 32, 169–176. [Google Scholar] [CrossRef] [PubMed]
- ArmaGen’s AGT-181 Demonstrates Neurocognitive Benefit in Children with Severe MPS I. Available online: http://armagen.com/news/armagens-agt-181-demonstrates-neurocognitive-benefit-children-severe-mps/ (accessed on 12 December 2019).
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol. Pharm. 2014, 11, 2928–2934. [Google Scholar] [CrossRef]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Lin, H.; Pardridge, W.M. Insulin Receptor Antibody-alpha-N-Acetylglucosaminidase Fusion Protein Penetrates the Primate Blood-Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type B Fibroblasts. Mol. Pharm. 2016, 13, 1385–1392. [Google Scholar] [CrossRef]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Ko, A.R.; Seong, M.R.; Lee, S.; Kim, M.R.; Cho, S.Y.; Kim, J.S.; Sakaguchi, M.; Nakazawa, T.; Kosuga, M.; et al. The efficacy of intracerebroventricular idursulfase-beta enzyme replacement therapy in mucopolysaccharidosis II murine model: Heparan sulfate in cerebrospinal fluid as a clinical biomarker of neuropathology. J. Inherit. Metab. Dis. 2018, 41, 1235–1246. [Google Scholar] [CrossRef]
- Safety and Dose Ranging Study of Human Insulin Receptor MAb-IDUA Fusion Protein in Adults and Children with MPS I. Available online: https://ClinicalTrials.gov/show/NCT03053089 (accessed on 12 December 2019).
- A Study to Test the Possibility of Cross Reaction Induced by the Idursulfase Drug to GSK2788723. Available online: https://ClinicalTrials.gov/show/NCT01602601 (accessed on 12 December 2019).
- A Study of JR-141 in Patients with Mucopolysaccharidosis II. Available online: https://ClinicalTrials.gov/show/NCT03359213 (accessed on 12 December 2019).
- Schmidt, M.; Boado, R.; Giugliani, R.; Pardridge, W. Anti-drug antibody response in mucopolysaccharidosis type I patients treated with AGT-181, a brain penetrating human insulin receptor antibody-iduronidase fusion protein. Mol. Genet. Metab. 2018, 123, S126. [Google Scholar] [CrossRef]
- Clinigen, K.K. and GC Pharma Announce Exclusive Licensing Agreement in Japan for Hunterase (Idursulfase-Beta ICV, Hunter Syndrome Drug). Available online: https://www.prnewswire.com/news-releases/clinigen-kk-and-gc-pharma-announce-exclusive-licensing-agreement-in-japan-for-hunterase-idursulfase-beta-icv-hunter-syndrome-drug-300824455.html (accessed on 12 December 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
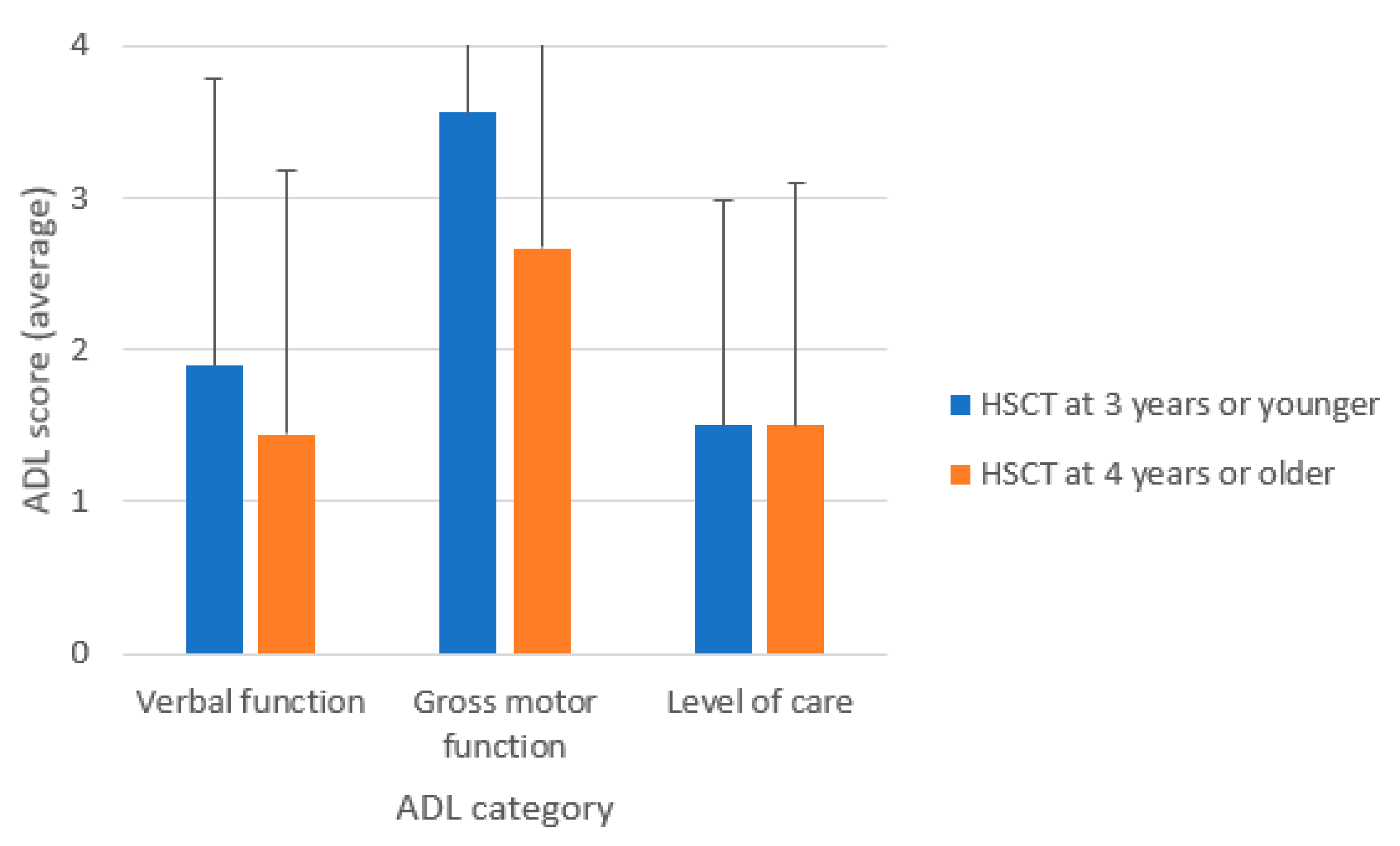{kind=link}
| Severe Phenotype | Attenuated Phenotype | |||
|---|---|---|---|---|
| Number of patients (M/F) | 69 (68/1) | 40 (40/0) | ||
| Age at onset (year) | 1.68 ± 1.30 ** | 3.08 ± 2.28 | ||
| Age at diagnosis (year) | 3.2 ± 1.80 ** | 7.08 ± 5.75 | ||
| Age at study (year) | 10.85 ± 5.86 ** | 16.69 ± 8.02 | ||
| HSCT | + | − | + | − |
| Number of patients | 19 | 50 | 5 | 35 |
| Age at onset (year) | 1.40 ± 1.06 | 1.87 ± 1.38 | 2.5 ± 2.18 | 3.14 ± 2.27 |
| Age at diagnosis (year) | 2.78 ± 1.77 | 3.36 ± 1.80 | 3.4 ± 2.07 | 7.64 ± 5.94 |
| Age in this study (year) | 10.89 ± 5.45 | 10.84 ± 6.07 | 13.0 ± 8.28 | 17.3 ± 7.95 |
| Age at HSCT | 4.11 ± 2.42 | 5.0 ± 2.74 | ||
| Clinical findings (%) | ||||
| Facial dysmorphism | 89 | 100 | 100 | 100 |
| Macroglossia | 59 ** | 90 | 0 ** | 68 |
| Frequent airway infection | 50 ** | 83 | 20 * | 76 |
| Coarse skin/hypertrichosis | 79 | 98 | 40 ** | 91 |
| Umbilical/inguinal hernia | 56 | 69 | 25 * | 81 |
| Hepatomegaly/splenomegaly | 78 * | 96 | 40 ** | 94 |
| Joint contracture | 84 * | 100 | 60 | 100 |
| Heart valve disorders | 63 | 80 | 60 * | 94 |
| Corneal clouding | 6 | 13 | 0 | 12 |
| Hearing difficulty | 67 | 85 | 60 | 77 |
| Sleep apnea | 11 ** | 63 | 0 | 35 |
| Central nervous symptoms | 89 | 98 | 40 | 47 |
| Carpal tunnel syndrome | 0 | 0 | 20 * | 16 |
| Dilated ventricle (MRI) | 47 | 71 | 40 | 39 |
| Category | 4 | 3 | 2 | 1 | 0 |
|---|---|---|---|---|---|
| Verbal function | Ordinary conversation | Two-words sentence | Single-word | Babbling | No word |
| Gross motor function | Ordinary walk | Walk with stick | Walk with aid | Wheelchair | Bed-ridden |
| Level of care | Independent | Moderately dependent | Highly dependent |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, Y.; Taylor, M.; Orii, K.; Fukao, T.; Orii, T.; Tomatsu, S. Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation. Diagnostics 2020, 10, 46. https://doi.org/10.3390/diagnostics10010046
Suzuki Y, Taylor M, Orii K, Fukao T, Orii T, Tomatsu S. Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation. Diagnostics. 2020; 10(1):46. https://doi.org/10.3390/diagnostics10010046
Chicago/Turabian StyleSuzuki, Yasuyuki, Madeleine Taylor, Kenji Orii, Toshiyuki Fukao, Tadao Orii, and Shunji Tomatsu. 2020. "Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation" Diagnostics 10, no. 1: 46. https://doi.org/10.3390/diagnostics10010046
APA StyleSuzuki, Y., Taylor, M., Orii, K., Fukao, T., Orii, T., & Tomatsu, S. (2020). Assessment of Activity of Daily Life in Mucopolysaccharidosis Type II Patients with Hematopoietic Stem Cell Transplantation. Diagnostics, 10(1), 46. https://doi.org/10.3390/diagnostics10010046