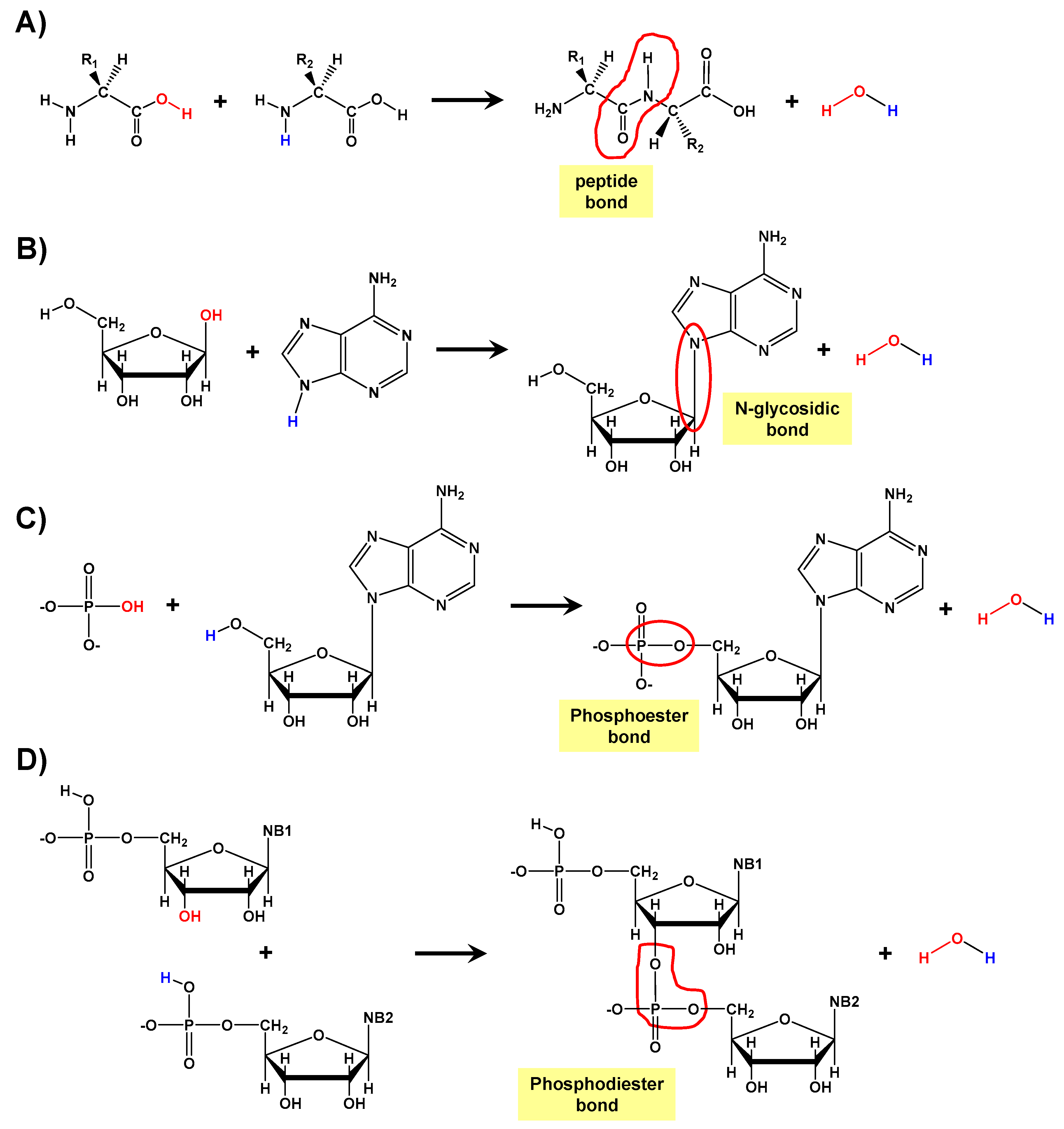
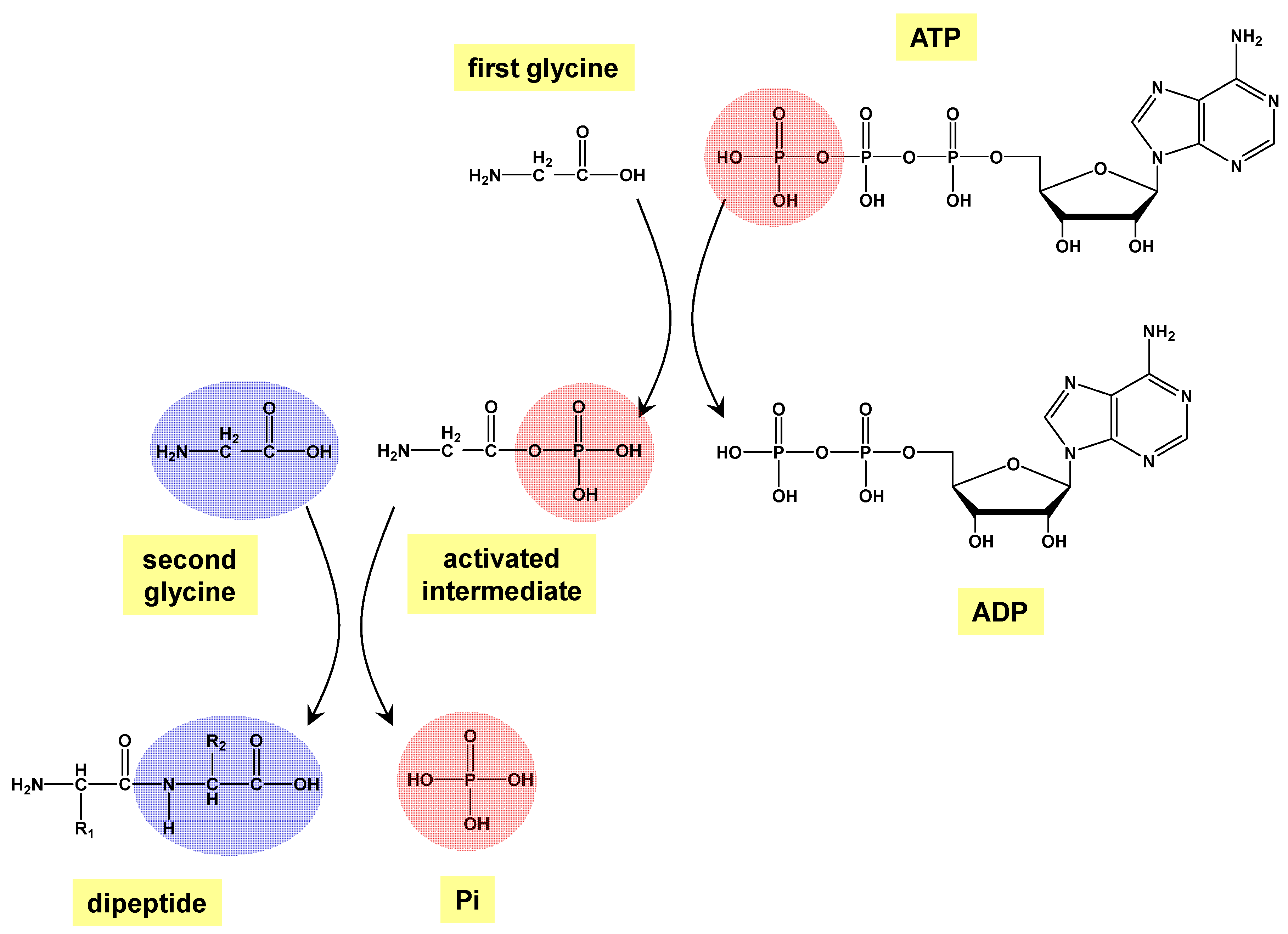
Prebiotic Peptide Bond Formation Through Amino Acid Phosphorylation. Insights from Quantum Chemical Simulations



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
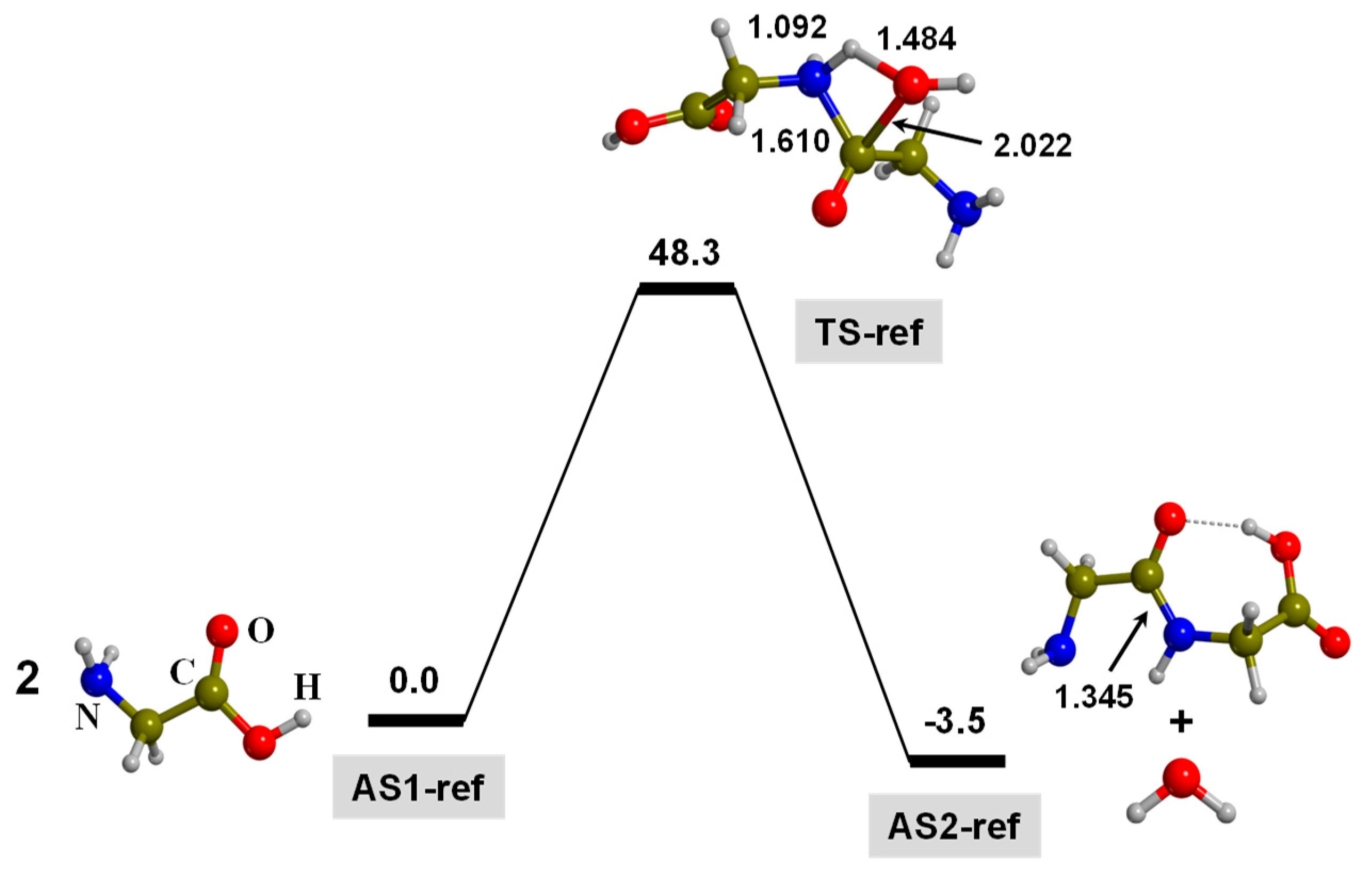
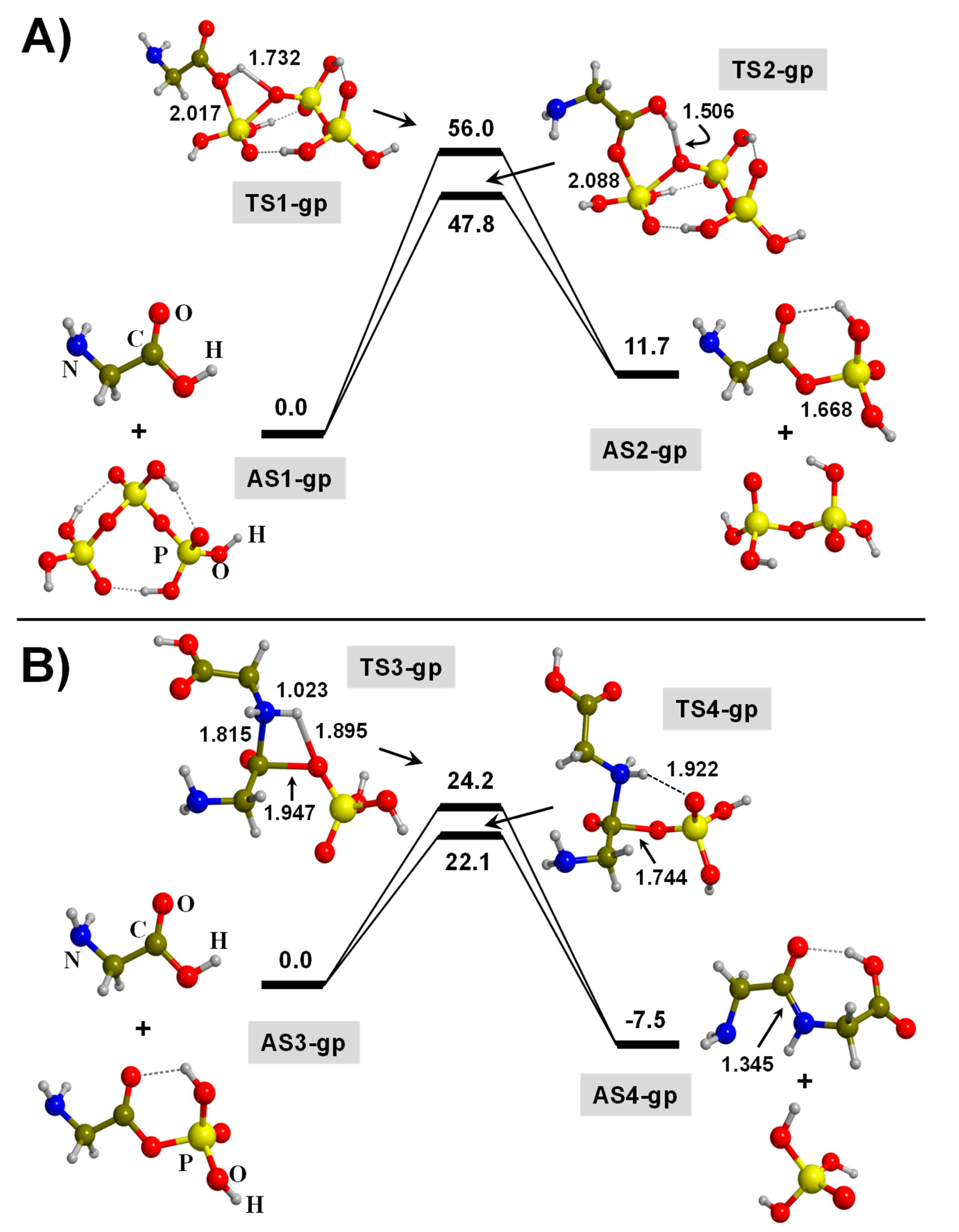
3. Results and Discussion
3.1. TP-Mediated Peptide Bond Formation in the Gas Phase
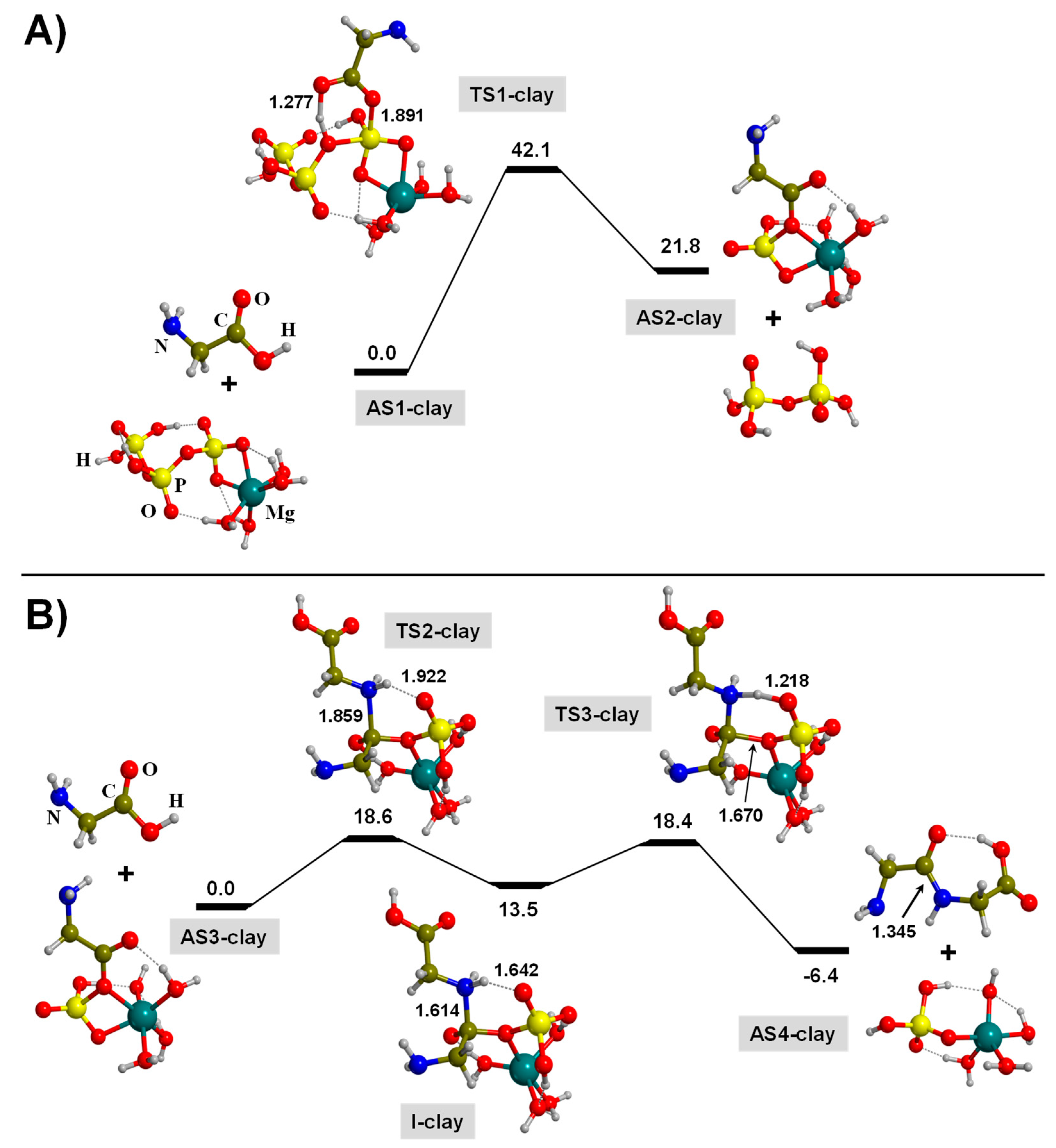
3.2. TP-Mediated Peptide Bond Formation in the Presence of A Mg2+-Clay Cation
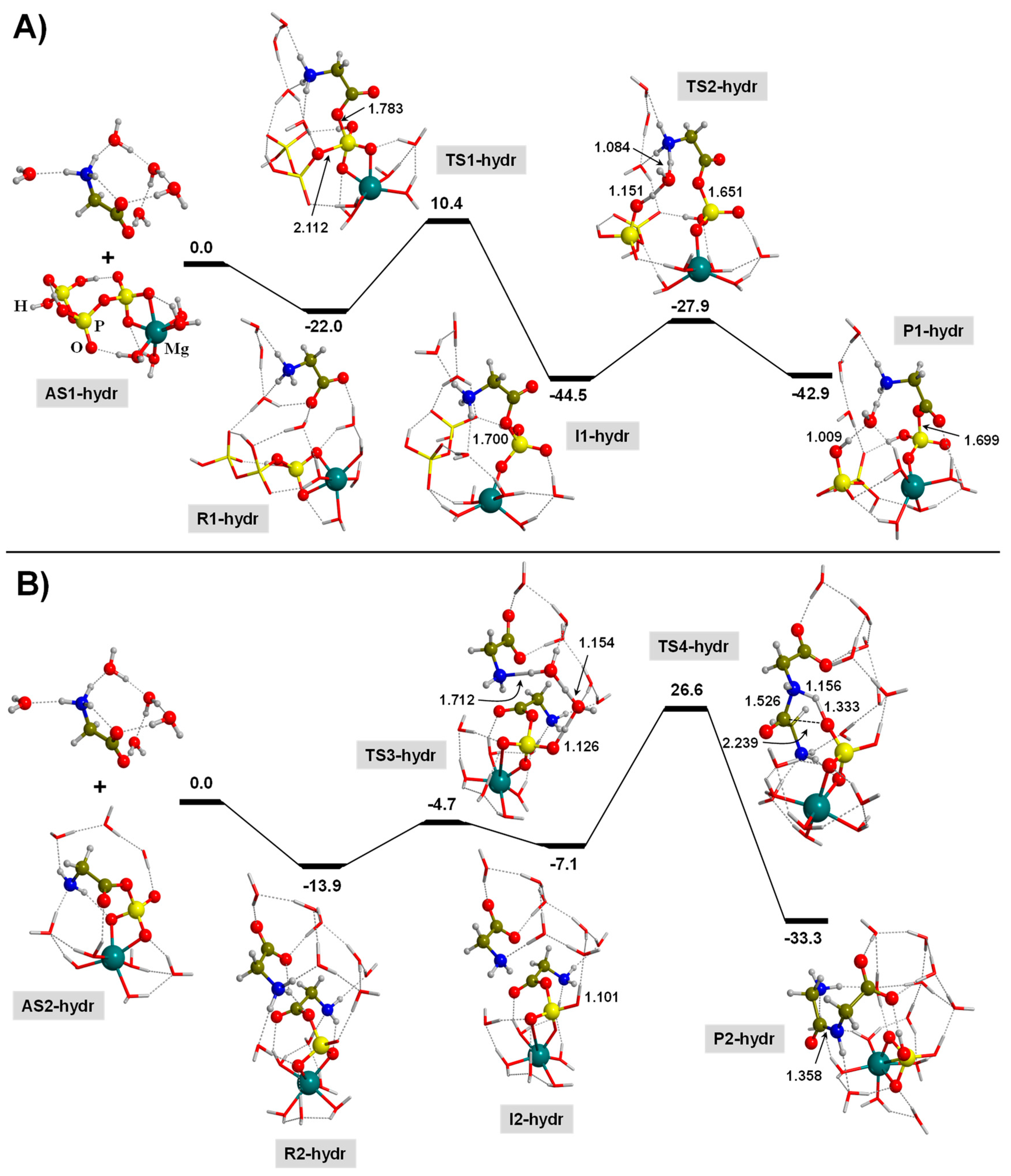
3.3. TP-Mediated Peptide Bond Formation in the Presence of a Mg2+ ion and in Watery Environments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hazen, R.M. Genesis: The Scientific Quest for Lifes Origin; The National Academies Press: Washington, DC, USA, 2005. [Google Scholar]
- Lehn, J.-M. Perspectives in Chemistry-Steps towards Complex Matter. Angew. Chem. Int. Ed. 2013, 52, 2836–2850. [Google Scholar] [CrossRef]
- Lehn, J.-M. Perspectives in Chemistry-Aspects of Adaptive Chemistry and Materials. Angew. Chem. Int. Ed. 2015, 54, 3276–3289. [Google Scholar] [CrossRef] [PubMed]
- Stano, P.; Luisi, P.L. Basic Questions About the Origins of Life: Proceedings of the Erice International School of Complexity (Fourth Course). Orig. Life Evol. Biosph. 2007, 37, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Brack, A. From Interstellar Amino Acids to Prebiotic Catalytic Peptides: A Review. Chem. Biodivers. 2007, 38, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. The origin of life—A review of facts and speculations. Trends Biochem. Sci. 1998, 23, 491–495. [Google Scholar] [CrossRef]
- Orgel, L.E. Prebiotic Chemistry and the Origin of the RNA World. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [PubMed]
- Saladino, R.; Crestini, C.; Ciciriello, F.; Costanzo, G.; Di Mauro, E. Formamide Chemistry and the Origin of Informational Polymers. Chem. Biodivers. 2007, 38, 694–720. [Google Scholar] [CrossRef]
- Lambert, J.-F. Adsorption and Polymerization of Amino Acids on Mineral Surfaces: A Review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef]
- Shock, E.L. Stability of peptides in high-temperature aqueous solutions. Geochim. Et Cosmochim. Acta 1992, 56, 3481–3491. [Google Scholar] [CrossRef]
- Benner, S.A. Paradoxes in the Origin of Life. Orig. Life Evol. Biosph. 2014, 44, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Westall, F.; Brack, A. The Importance of Water for Life. Space Sci. Rev. 2018, 214, 50. [Google Scholar] [CrossRef]
- Bernal, J.D. The Physical Basis of Life. Proc. Phys. Soc. B 1949, 62, 597. [Google Scholar] [CrossRef]
- Orgel, L.E. Polymerization on the Rocks: Theoretical Introduction. Orig. Life Evol. Biosph. 1998, 28, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.V. Biochemical evolution. I. Polymerization on internal, organophilic silica surfaces of dealuminated zeolites and feldspars. Proc. Natl. Acad. Sci. USA 1998, 95, 3370–3375. [Google Scholar] [CrossRef] [PubMed]
- Lahav, N.; White, D.; Chang, S. Peptide formation in the prebiotic era: Thermal condensation of glycine in fluctuating clay environments. Science 1978, 201, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.F.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Genet. 2008, 6, 805–814. [Google Scholar] [CrossRef]
- Iqubal, M.A.; Sharma, R.; Jheeta, S. Kamaluddin Thermal Condensation of Glycine and Alanine on Metal Ferrite Surface: Primitive Peptide Bond Formation Scenario. Life 2017, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Jheeta, S. The Routes of Emergence of Life from LUCA during the RNA and Viral World: A Conspectus. Life 2015, 5, 1445–1453. [Google Scholar] [CrossRef]
- Lambert, J.-F.; Jaber, M.; Georgelin, T.; Stievano, L. A comparative study of the catalysis of peptide bond formation by oxide surfaces. Phys. Chem. Chem. Phys. 2013, 15, 13371–13380. [Google Scholar] [CrossRef]
- Martra, G.; Deiana, C.; Sakhno, Y.; Barberis, I.; Fabbiani, M.; Pazzi, M.; Vincenti, M. The Formation and Self-Assembly of Long Prebiotic Oligomers Produced by the Condensation of Unactivated Amino Acids on Oxide Surfaces. Angew. Chem. 2014, 126, 4759–4762. [Google Scholar] [CrossRef]
- Rimola, A.; Fabbiani, M.; Sodupe, M.; Ugliengo, P.; Martra, G. How Does Silica Catalyze the Amide Bond Formation under Dry Conditions? Role of Specific Surface Silanol Pairs. ACS Catal. 2018, 8, 4558–4568. [Google Scholar] [CrossRef]
- Deiana, C.; Sakhno, Y.; Fabbiani, M.; Pazzi, M.; Vincenti, M.; Martra, G. Direct Synthesis of Amides from Carboxylic Acids and Amines by Using Heterogeneous Catalysts: Evidence of Surface Carboxylates as Activated Electrophilic Species. ChemCatChem 2013, 5, 2832–2834. [Google Scholar] [CrossRef]
- Barcaro, G.; Sementa, L.; Carravetta, V.; Yano, T.-A.; Hara, M.; Monti, S. Experimental and theoretical elucidation of catalytic pathways in TiO2-initiated prebiotic polymerization. Phys. Chem. Chem. Phys. 2019, 21, 5435–5447. [Google Scholar] [CrossRef]
- Bujdak, J.; Rode, B.M. Activated alumina as an energy source for peptide bond formation: Consequences for mineral-mediated prebiotic processes. Amino Acids 2001, 21, 281–291. [Google Scholar] [CrossRef]
- Bujdak, J.; Rode, B. Preferential amino acid sequences in alumina-catalyzed peptide bond formation. J. Inorg. Biochem. 2002, 90, 1–7. [Google Scholar] [CrossRef]
- Bujdak, J.; Rode, B. Peptide Bond Formation on the Surface of Activated Alumina: Peptide Chain Elongation. Catal. Lett. 2003, 91, 149–154. [Google Scholar] [CrossRef]
- Bujdak, J.; Rode, B.M. Alumina catalyzed reactions of amino acids. J. Therm. Anal. Calorim 2003, 73, 797–805. [Google Scholar] [CrossRef]
- Ferris, J.P.; Hill, A.R.; Liu, R.; Orgel, L.E. Synthesis of long prebiotic oligomers on mineral surfaces. Nature 1996, 381, 59–61. [Google Scholar] [CrossRef]
- Georgelin, T.; Akouche, M.; Jaber, M.; Sakhno, Y.; Matheron, L.; Fournier, F.; Méthivier, C.; Martra, G.; Lambert, J.-F. Iron (III) Oxide Nanoparticles as Catalysts for the Formation of Linear Glycine Peptides. Eur. J. Inorg. Chem. 2017, 1, 198–211. [Google Scholar] [CrossRef]
- Gregoire, B.; Greenwell, H.C.; Fraser, D.G. Peptide Formation on Layered Mineral Surfaces: The Key Role of Brucite-like Minerals on the Enhanced Formation of Alanine Dipeptides. ACS Earth Space Chem. 2018, 2, 852–862. [Google Scholar] [CrossRef]
- Matrajt, G.; Blanot, D. Properties of synthetic ferrihydrite as an amino acid adsorbent and a promoter of peptide bond formation. Amino Acids 2004, 26, 153–158. [Google Scholar] [CrossRef]
- Kitadai, N.; Oonishi, H.; Umemoto, K.; Usui, T.; Fukushi, K.; Nakashima, S. Glycine Polymerization on Oxide Minerals. Orig. Life Evol. Biosph. 2017, 47, 123–143. [Google Scholar] [CrossRef]
- Ferris James, P. Montmorillonite-catalysed formation of RNA oligomers: The possible role of catalysis in the origins of life. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1777–1786. [Google Scholar] [CrossRef]
- Rimola, A.; Sodupe, M.; Ugliengo, P. Role of Mineral Surfaces in Prebiotic Chemical Evolution. In Silico Quantum Mechanical Studies. Life 2019, 9, 10. [Google Scholar] [CrossRef]
- Cleaves, H.J., II; Scott, A.M.; Hill, F.C.; Leszczynski, J.; Sahai, N.; Hazen, R. Mineral–organic interfacial processes: Potential roles in the origins of life. Chem. Soc. Rev. 2012, 41, 5502–5525. [Google Scholar] [CrossRef]
- Pérez-Villa, A.; Pietrucci, F.; Saitta, A.M. Prebiotic chemistry and origins of life research with atomistic computer simulations. Phys. Life Rev. 2018. [Google Scholar] [CrossRef]
- Ugliengo, P. The rise of computer modeling in prebiotic chemistry: Comment on “Prebiotic chemistry and origins of life research with atomistic computer simulations” by A. Pérez-Villa et al. Phys. Life Rev. 2019. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of Potentially Prebiotic Condensed Phosphates by Phosphorus Redox Chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef]
- Qasim, D.; Abbott-Lyon, H.; Pirim, C.; Orlando, T.; Gull, M.; Lindsay, D.; La Cruz, N.L.; McKee, A.D.; Pasek, M.A. The evolution of the surface of the mineral schreibersite in prebiotic chemistry. Phys. Chem. Chem. Phys. 2016, 18, 20160–20167. [Google Scholar]
- Akouche, M.; Jaber, M.; Maurel, M.-C.; Lambert, J.-F.; Georgelin, T. Phosphoribosyl Pyrophosphate: A Molecular Vestige of the Origin of Life on Minerals. Angew. Chem. Int. Ed. 2017, 56, 7920–7923. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef]
- Coggins, A.J.; Powner, M.W. Prebiotic synthesis of phosphoenol pyruvate by α-phosphorylation-controlled triose glycolysis. Nat. Chem. 2016, 9, 310–317. [Google Scholar] [CrossRef]
- Gull, M.; Mojica, M.A.; Fernández, F.M.; Gaul, D.A.; Orlando, T.M.; Liotta, C.L.; Pasek, M.A. Nucleoside phosphorylation by the mineral schreibersite. Sci. Rep. 2015, 5, 17198. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Antony, J.; Krieg, H.; Grimme, S.; Ehrlich, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- McQuarrie, D. Statistical Mechanics; Harper and Row: New York, NY, USA, 1986. [Google Scholar]
- Ramaekers, R.; Pająk, J.; Lambie, B.; Maes, G. Neutral and zwitterionic glycine.H2O complexes: A theoretical and matrix-isolation Fourier transform infrared study. J. Chem. Phys. 2004, 120, 4182. [Google Scholar] [CrossRef]
- Blom, M.N.; Compagnon, I.; Polfer, N.C.; Von Helden, G.; Meijer, G.; Suhai, S.; Paizs, B.; Oomens, J. Stepwise Solvation of an Amino Acid: The Appearance of Zwitterionic Structures. J. Phys. Chem. A 2007, 111, 7309–7316. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Bachs, B.; Rimola, A. Prebiotic Peptide Bond Formation Through Amino Acid Phosphorylation. Insights from Quantum Chemical Simulations. Life 2019, 9, 75. https://doi.org/10.3390/life9030075
Martínez-Bachs B, Rimola A. Prebiotic Peptide Bond Formation Through Amino Acid Phosphorylation. Insights from Quantum Chemical Simulations. Life. 2019; 9(3):75. https://doi.org/10.3390/life9030075
Chicago/Turabian StyleMartínez-Bachs, Berta, and Albert Rimola. 2019. "Prebiotic Peptide Bond Formation Through Amino Acid Phosphorylation. Insights from Quantum Chemical Simulations" Life 9, no. 3: 75. https://doi.org/10.3390/life9030075
APA StyleMartínez-Bachs, B., & Rimola, A. (2019). Prebiotic Peptide Bond Formation Through Amino Acid Phosphorylation. Insights from Quantum Chemical Simulations. Life, 9(3), 75. https://doi.org/10.3390/life9030075