Wet-Dry Cycling Delays the Gelation of Hyperbranched Polyesters: Implications to the Origin of Life

Abstract
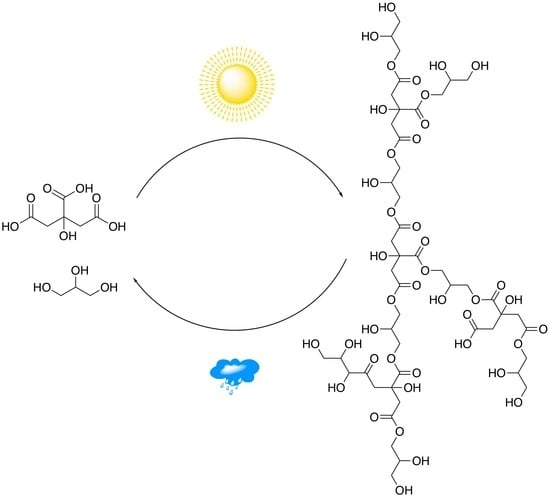
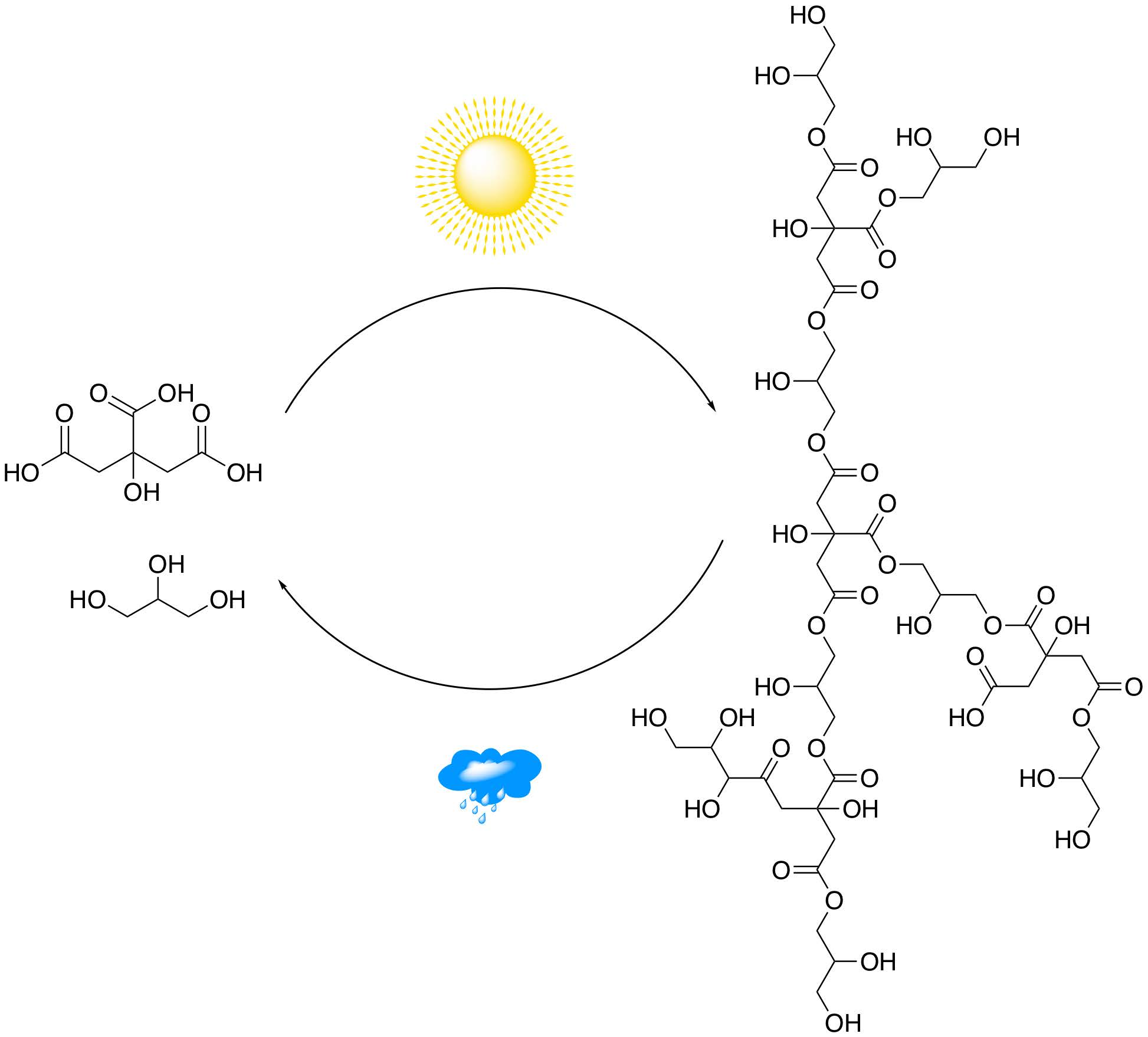
1. Introduction
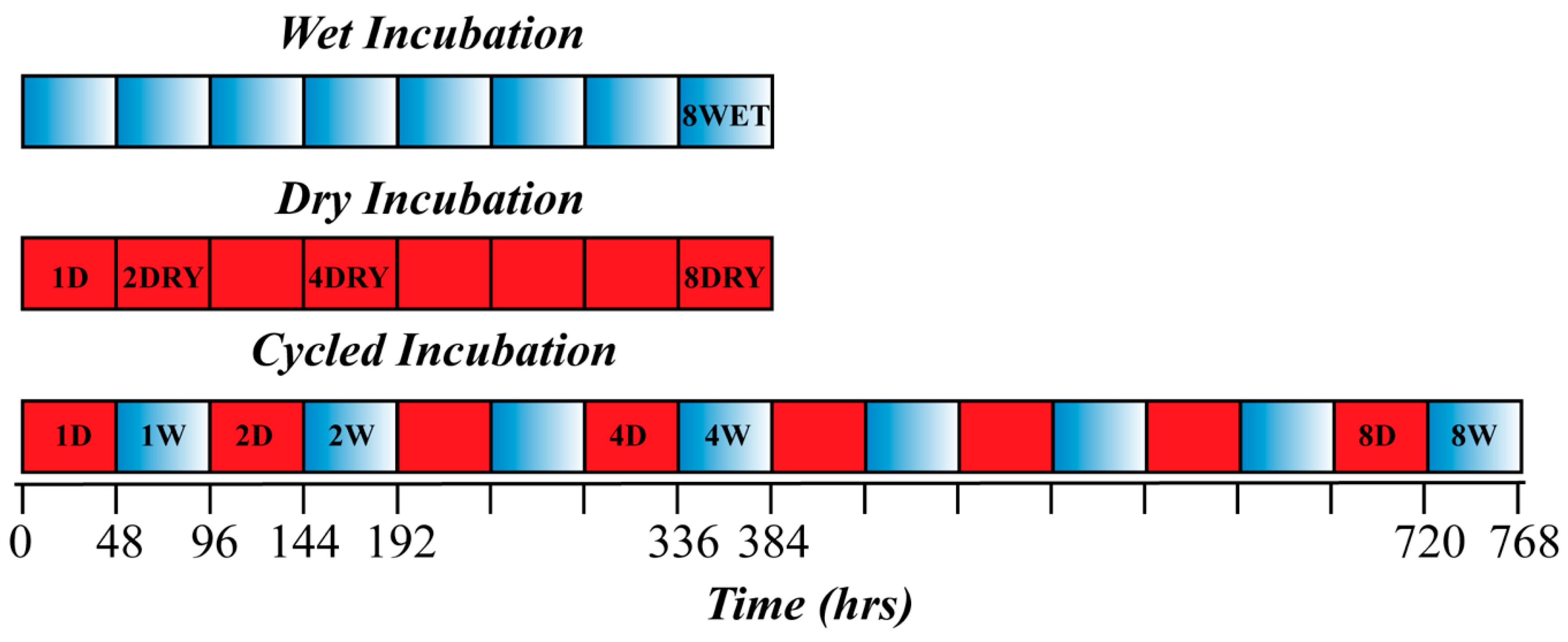
2. Materials and Methods
3. Results
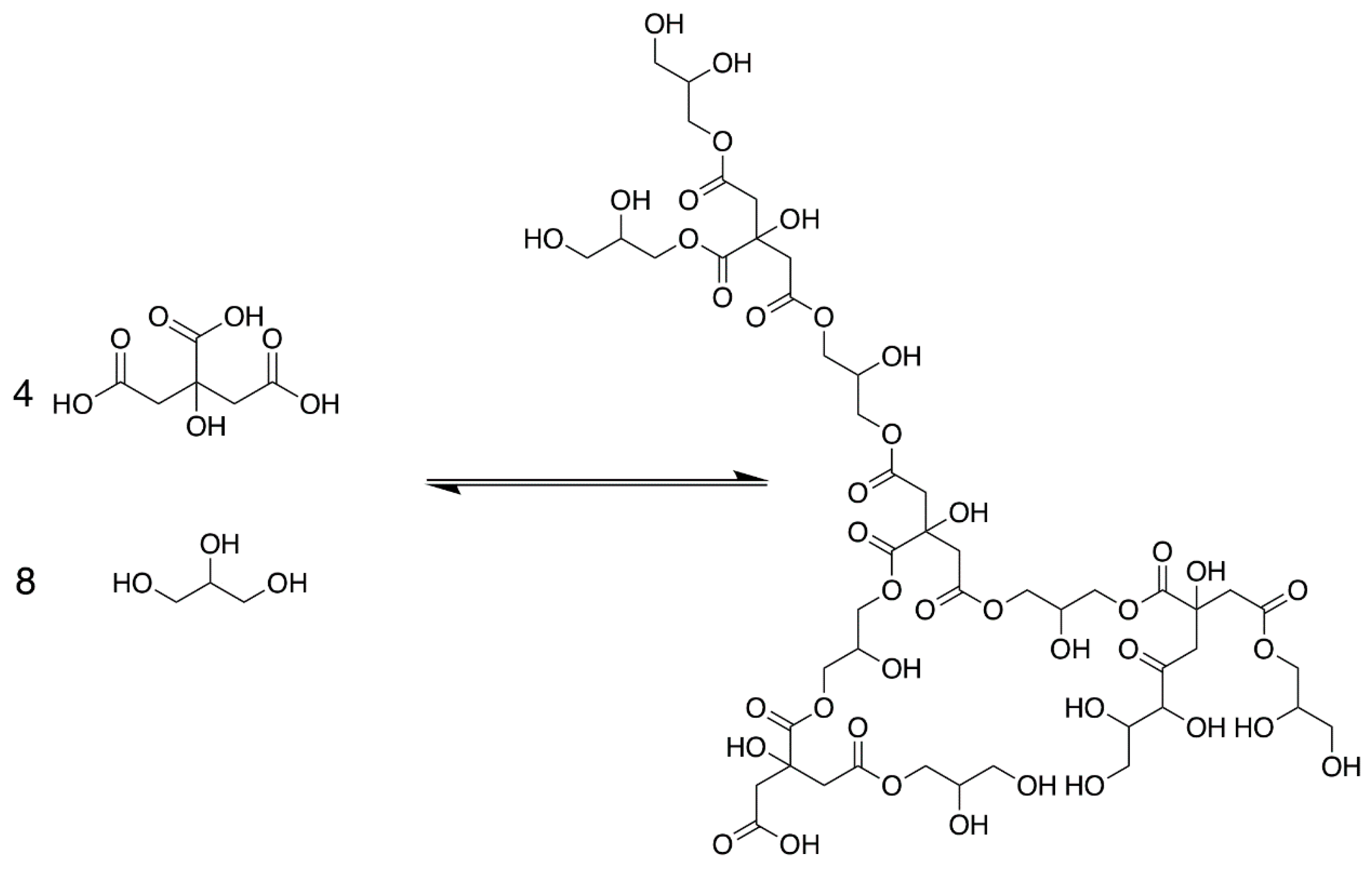
3.1. Polyesterification
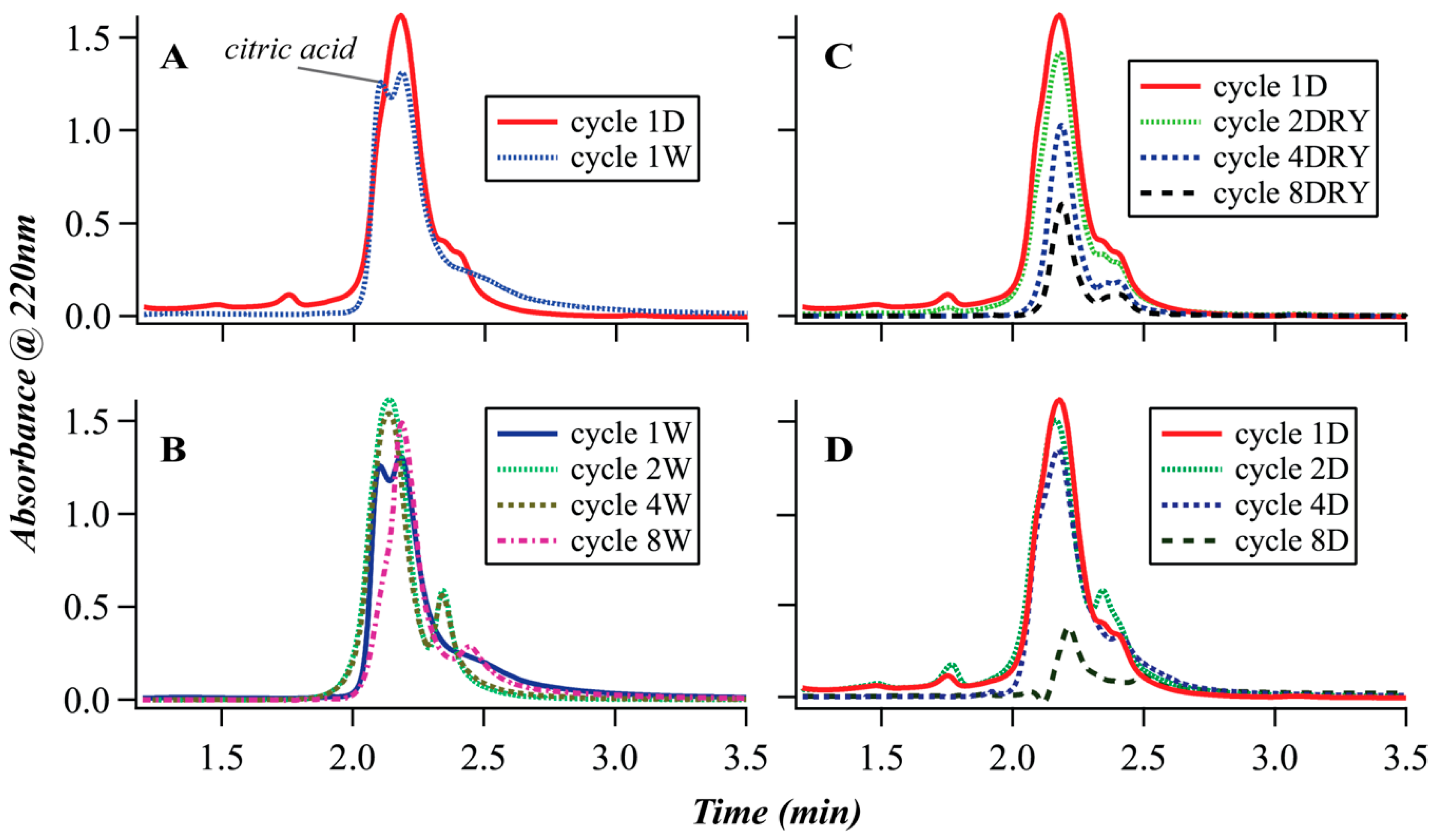
3.2. Size-Exclusion Chromatography (SEC)
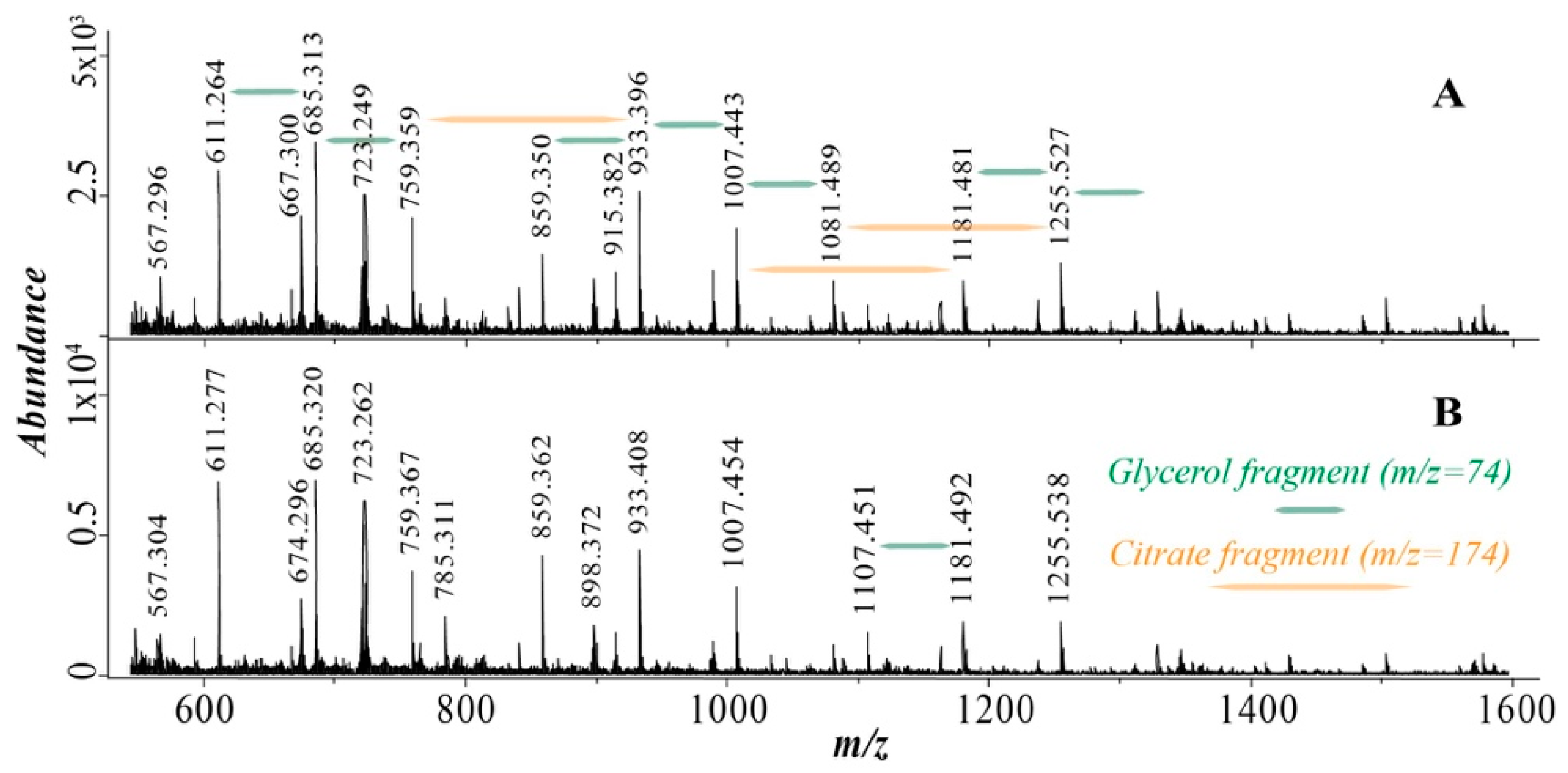
3.3. Mass Spectrometry
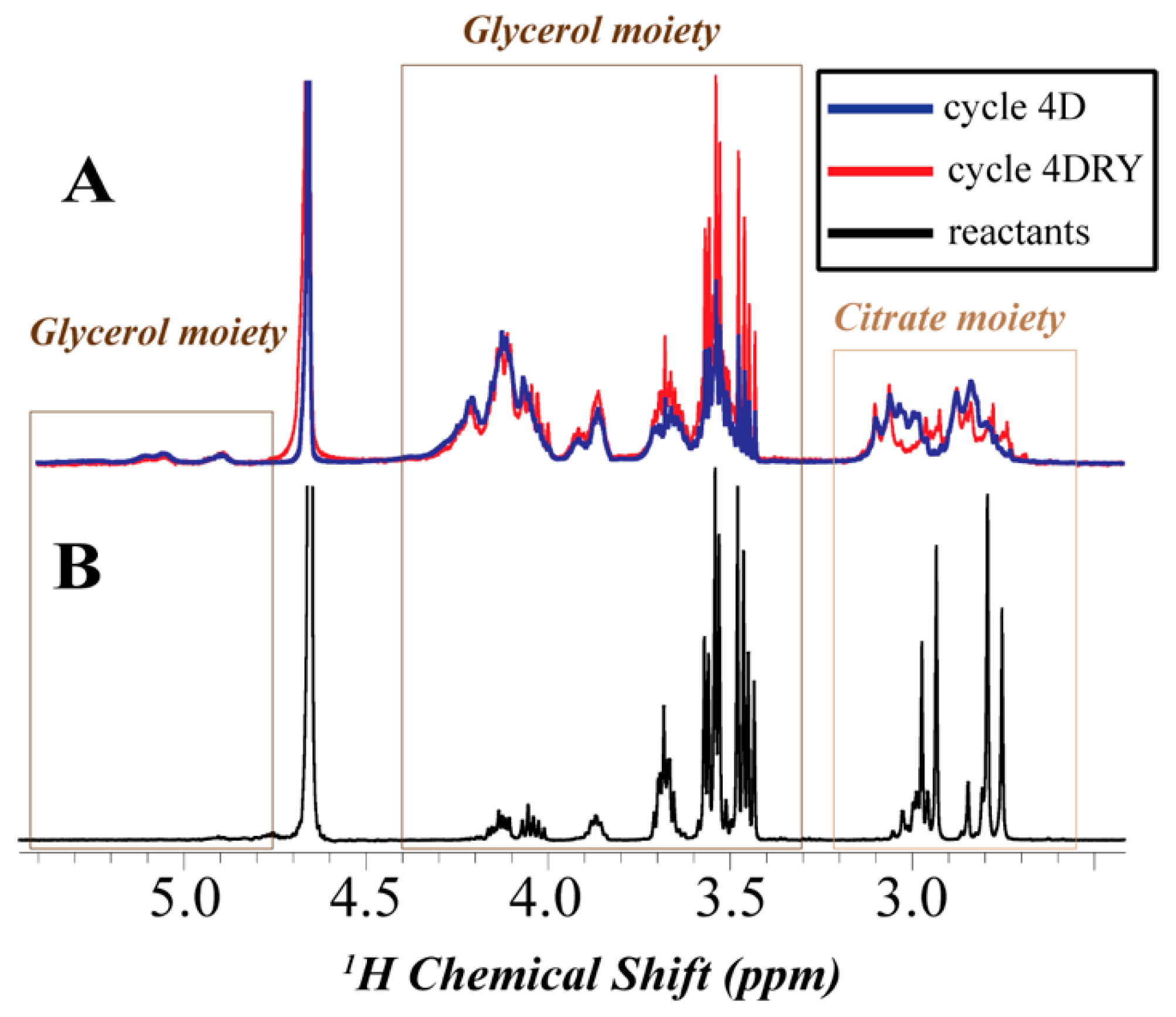
3.4. Nuclear Magnetic Resonance (NMR)
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 7th ed.; W.H. Freeman: New York, NY, USA, 2012. [Google Scholar]
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Urey, H.C. Organic Compound Synthes on the Primitive Earth: Several questions about the origin of life have been answered, but much remains to be studied. Science 1959, 130, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.R.; Moore, C.B. Amino Acid Analyses of the Murchison, Murray, and Allende Carbonaceous Chondrites. Science 1971, 172, 1327–1329. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.R.; Pizzarello, S. Amino acids in meteorites. Adv. Space Res. 1983, 3, 5–18. [Google Scholar] [CrossRef]
- Ehrenfreund, P.; Glavin, D.P.; Botta, O.; Cooper, G.; Bada, J.L. Extraterrestrial Amino Acids in Orgueil and Ivuna: Tracing the Parent Body of CI-type Carbonaceous Chondrites. Proc. Natl. Acad. Sci. USA 2001, 98, 2138–2141. [Google Scholar] [CrossRef] [PubMed]
- Oró, J. Mechanism of Synthesis of Adenine from Hydrogen Cyanide under Possible Primitive Earth Conditions. Nature 1961, 191, 1193–1194. [Google Scholar] [CrossRef]
- Barks, H.L.; Buckley, R.; Grieves, G.A.; Di Mauro, E.; Hud, N.V.; Orlando, T.M. Guanine, Adenine, and Hypoxanthine Production in UV-irradiated Formamide Solutions: Relaxation of the Requirements for Prebiotic Purine Nucleobase Formation. Chembiochem. Eur. J. Chem. Biol. 2010, 11, 1240–1243. [Google Scholar] [CrossRef]
- Breslow, R. On the Mechanism of the Formose Reaction. Tetrahedron Lett. 1959, 1, 22–26. [Google Scholar] [CrossRef]
- Ricardo, A.; Frye, F.; Carrigan, M.A.; Tipton, J.D.; Powell, D.H.; Benner, S.A. 2-Hydroxymethylboronate as a Reagent to Detect Carbohydrates: Application to the Analysis of the Formose Reaction. J. Org. Chem. 2006, 71, 9503–9505. [Google Scholar] [CrossRef]
- Pizzarello, S.; Shock, E. The Organic Composition of Carbonaceous Meteorites: The Evolutionary Story Ahead of Biochemistry. Cold Spring Harb. Perspect. Biol. 2010, 2, a002105. [Google Scholar] [CrossRef]
- Forsythe, J.G.; Yu, S.S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernández, F.M.; Hud, N.V. Ester-Mediated Amide Bond Formation Driven by Wet–Dry Cycles: A Possible Path to Polypeptides on the Prebiotic Earth. Angew. Chem. Int. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.T.; Suárez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of Oligopeptides in High Yield under Simple Programmable Conditions. Nat. Commun. 2015, 6, 8385. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Hill, A.R.; Liu, R.; Orgel, L.E. Synthesis of Long Prebiotic Oligomers on Mineral Surfaces. Nature 1996, 381, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Olasagasti, F.; Deamer, D. Lipid-assisted Synthesis of RNA-like Polymers from Mononucleotides. Orig. Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Bernloehr, H.; Lohrmann, R.; Sulston, J.; Weimann, B.J.; Orgel, L.E.; Miles, H.T. Non-enzymic Synthesis of Deoxyadenylate Oligonucleotides on a Polyuridylate Template. J. Mol. Biol. 1968, 37, 151–155. [Google Scholar] [CrossRef]
- Eschenmoser, A. Searching for Nucleic Acid Alternatives. Chimia 2005, 59, 836–850. [Google Scholar] [CrossRef]
- Bean, H.D.; Anet, F.A.L.; Gould, I.R.; Hud, N.V. Glyoxylate as a Backbone Linkage for a Prebiotic Ancestor of RNA. Orig. Life Evol. Biosph. 2006, 36, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Mamajanov, I.; Cody, G.D. Protoenzymes: The Case of Hyperbranched Polyesters. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2017, 357, 20160357. [Google Scholar] [CrossRef]
- Mamajanov, I.; Herzfeld, J. HCN Polymer. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin, Germany, 2011; pp. 730–732. [Google Scholar]
- Benner, S.A. Paradoxes in the Origin of Life. Orig. Life Evol. Biosph. 2014, 44, 339–343. [Google Scholar] [CrossRef]
- Fox, S.W.; Krampitz, G. Catalytic Decomposition of Glucose in Aqueous Solution by Thermal Proteinoids. Nature 1964, 203, 1362–1364. [Google Scholar] [CrossRef]
- Durant, D.; Fox, S. Enhancement of Rate of Decarboxylation of Pyruvic Acid and of Hydrolysis. Fed. Proc. 1966, 25, 342–346. [Google Scholar]
- Rohlfing, D.; Fox, S. Catalytic Activity of Thermal Polyanhydro-Alpha-Amino Acids for Hydrolysis of P-Nitrophenyl Acetate—Catalysis by Thermal Polyamino Acids. Arch. Biochem. Biophys. 1967, 118, 122–126. [Google Scholar] [CrossRef]
- Flory, P.J. Fundamental Principles of Condensation Polymerization. Chem. Rev. 1946, 39, 137–197. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.B. Free Energies and Equilibria of Peptide Bond Hydrolysis and Formation. Biopolymers 1998, 45, 351–353. [Google Scholar] [CrossRef]
- Williams, R.J.; Gabriel, A.; Andrews, R.C. The Relation Between the Hydrolysis Equilibrium Constant of Esters and the Strengths of the Corresponding Acids. J. Am. Chem. Soc. 1928, 50, 1267–1271. [Google Scholar] [CrossRef]
- Orgel, L.E. Some Consequences of the RNA World Hypothesis. Orig. Life Evol. Biosph. 2003, 33, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, S.; Neumann, H.; Shashoua, V.; Rich, A. Ribosome-catalyzed ester formation. Biochemistry 1970, 9, 2477–2483. [Google Scholar] [CrossRef]
- Fahnestock, S.; Rich, A. Ribosome-Catalyzed Polyester Formation. Science 1971, 173, 340–343. [Google Scholar] [CrossRef]
- Weber, A.L. Thermal Synthesis and Hydrolysis of Polyglyceric Acid. Orig. Life Evol. Biosph. 1989, 19, 7–19. [Google Scholar] [CrossRef]
- Mamajanov, I.; MacDonald, P.J.; Ying, J.; Duncanson, D.M.; Dowdy, G.R.; Walker, C.A.; Engelhart, A.E.; Fernandez, F.M.; Grover, M.A.; Hud, N.V.; et al. Ester Formation and Hydrolysis during Wet–Dry Cycles: Generation of Far-from-Equilibrium Polymers in a Model Prebiotic Reaction. Macromolecules 2014, 47, 1334–1343. [Google Scholar] [CrossRef]
- Medronho, B.; Romano, A.; Miguel, M.G.; Stigsson, L.; Lindman, B. Rationalizing Cellulose (In) solubility: Reviewing Basic Physicochemical Aspects and Role of Hydrophobic Interactions. Cellulose 2012, 19, 581–587. [Google Scholar] [CrossRef]
- Krässig, H.; Kitchen, W. Factors Influencing Tensile Properties of Cellulose Fibers. J. Polym. Sci. 1961, 51, 123–172. [Google Scholar] [CrossRef]
- Kirkorian, K.; Ellis, A.; Twyman, L.J. Catalytic Hyperbranched Polymers as Enzyme Mimics; Exploiting the Principles of Encapsulation and Supramolecular chemistry. Chem. Soc. Rev. 2012, 41, 6138–6159. [Google Scholar] [CrossRef]
- Astruc, D.; Chardac, F. Dendritic Catalysts and Dendrimers in Catalysis. Chem. Rev. 2001, 101, 2991–3024. [Google Scholar] [CrossRef] [PubMed]
- Bhyrappa, P.; Young, J.K.; Moore, J.S.; Suslick, K.S. Dendrimer-Metalloporphyrins: Synthesis and Catalysis. J. Am. Chem. Soc. 1996, 118, 5708–5711. [Google Scholar] [CrossRef]
- Liu, L.; Breslow, R. Dendrimeric Pyridoxamine Enzyme Mimics. J. Am. Chem. Soc. 2003, 125, 12110–12111. [Google Scholar] [CrossRef] [PubMed]
- Tasaka, F.; Ohya, Y.; Ouchi, T. One-Pot Synthesis of Novel Branched Polylactide Through the Copolymerization of Lactide with Mevalonolactone. Macromol. Rapid Commun. 2001, 22, 820–824. [Google Scholar] [CrossRef]
- Kumar, A.; Ramakrishnan, S. A Novel One-Pot Synthesis of Hyperbranched Polyurethanes. J. Chem. Soc. Chem. Commun. 1993, 1453–1454. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Hawker, C.J; Frechet, J.M.J.; Turner, S.R. One-Pot Synthesis of Hyperbranched Polyethers. Macromolecules 1992, 25, 4583–4587. [Google Scholar] [CrossRef]
- Mamajanov, I.; Callahan, M.P.; Dworkin, J.P.; Cody, G.D. Prebiotic Alternatives to Proteins: Structure and Function of Hyperbranched Polyesters. Orig. Life Evol. Biosph. 2015, 45, 123–137. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. VI. Branched Polymers Containing A-R-Bf-1 Type Units. J. Am. Chem. Soc. 1952, 74, 2718–2723. [Google Scholar] [CrossRef]
- Fréchet, J.M.J.; Hawker, C.J. Hyperbranched Polyphenylene and Hyperbranched Polyesters: New Soluble, Three-Dimensional, Reactive Polymers. React. Funct. Polym. 1995, 26, 127–136. [Google Scholar] [CrossRef]
- Kim, Y.H.; Webster, O.W. Water Soluble Hyperbranched Polyphenylene: “A Unimolecular Micelle? J. Am. Chem. Soc. 1990, 112, 4592–4593. [Google Scholar] [CrossRef]
- Wooley, K.L.; Hawker, C.J.; Lee, R.; Fréchet, J.M.J. One-Step Synthesis of Hyperbranched Polyesters. Molecular Weight Control and Chain End Functionalization. Polym. J. 1994, 26, 187–197. [Google Scholar] [CrossRef]
- Malmström, E.; Johansson, M.; Hult, A. Hyperbranched Aliphatic Polyesters. Macromolecules 1995, 28, 1698–1703. [Google Scholar] [CrossRef]
- Malmström, E.; Hult, A. Kinetics of Formation of Hyperbranched Polyesters Based on 2,2-Bis(methylol)propionic Acid. Macromolecules 1996, 29, 1222–1228. [Google Scholar] [CrossRef]
- Gao, C.; Yan, D. Hyperbranched Polymers: From Synthesis to Applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation1. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar] [CrossRef]
- Halpern, J.M.; Urbanski, R.; Weinstock, A.K.; Iwig, D.F.; Mathers, R.T.; von Recum, H.A. A Biodegradable Thermoset Polymer Made by Esterification of Citric Acid and Glycerol. J. Biomed. Mater. Res. A 2014, 102, 1467–1477. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, Y.; Roberts, E.; Duxbury, C.J.; Ding, L.; Irvine, D.J.; Howdle, S.M. Controlling Chain Growth: A New Strategy to Hyperbranched Materials. Macromolecules 2007, 40, 7184–7194. [Google Scholar] [CrossRef]
- Wang, M.; Gan, D.; Wooley, K.L. Linear and Hyperbranched Poly[silyl ester]s: Synthesis via Cross-Dehydrocoupling-Based Polymerization, Hydrolytic Degradation Properties, and Morphological Analysis by Atomic Force Microscopy. Macromolecules 2001, 34, 3215–3223. [Google Scholar] [CrossRef]
- Wu, D.; Liu, Y.; Jiang, X.; He, C.; Goh, S.H.; Leong, K.W. Evaluation of Hyperbranched Poly[amino ester]s of Amine Constitutions Similar to Polyethylenimine for DNA Delivery. Biomacromolecules 2005, 6, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liu, Y.; Jiang, X.; He, C.; Goh, S.H.; Leong, K.W. Hyperbranched Poly[amino ester]s with Different Terminal Amine Groups for DNA Delivery. Biomacromolecules 2006, 7, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, A. Comparison of Properties among Dendritic and Hyperbranched Poly[ether ether ketone]s and Linear Poly[ether ketone]s. Molecules 2016, 21, 219. [Google Scholar] [CrossRef] [PubMed]
- Newkome, G.R.; Young, J.K.; Baker, G.R.; Potter, R.L.; Audol, L.; Cooper, D.; Weis, C.D; Morris, K.; Johnson, C.S. Cascade Polymers. 35. pH Dependence of Hydrodynamic Radii of Acid-Terminated Dendrimers. Macromolecules 1993, 26, 2394–2396. [Google Scholar] [CrossRef]
- Cody, G.D.; Heying, E.; Alexander, C.M.O.; Nittler, L.R.; Kilcoyne, A.L.D.; Sandford, S.A.; Stroud, R.M. Establishing a Molecular Relationship Between Chondritic and Cometary Organic Solids. Proc. Natl. Acad. Sci. USA 2011, 108, 19171–19176. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Powner, M.W. Prebiotic Systems Chemistry: Complexity Overcoming Clutter. Chem 2017, 2, 470–501. [Google Scholar] [CrossRef]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic Systems Chemistry: New Perspectives for the origins of life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef]
- Guttenberg, N.; Virgo, N.; Chandru, K.; Scharf, C.; Mamajanov, I. Bulk Measurements of Messy Chemistries are Needed for a Theory of the Origins of Life. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2017, 375, 20160347. [Google Scholar] [CrossRef]
- Virgo, N.; Ikegami, T. Autocatalysis Before Enzymes: The Emergence of Prebiotic Chain Reactions. In Proceedings of the Artificial Life Conference Proceedings 13, Cambridge, MA, USA, July 2013; MIT Press: Cambridge, MA, USA, 2013; pp. 240–247. [Google Scholar]
- Virgo, N.; Ikegami, T.; McGregor, S. Complex Autocatalysis in Simple Chemistries. Artif. Life 2016, 22, 138–152. [Google Scholar] [CrossRef]
- Jain, S.; Krishna, S. Graph Theory and the Evolution of Autocatalytic Networks. In Handbook of Graphs and Networks; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2005; pp. 355–395. [Google Scholar]
- Drexler, K.E. Molecular Imprinting: The Missing Piece in the Puzzle of Abiogenesis? arXiv, 2018; 1807.07065v1. [Google Scholar]
- Wang, K.-J.; Ferris, J.P. Catalysis and Selectivity in Prebiotic Synthesis: Initiation of the Formation of Oligo[U]s on Montmorillonite Clay by Adenosine-5’-methylphosphate. Orig. Life Evol. Biosph. 2005, 35, 187–212. [Google Scholar] [CrossRef] [PubMed]
- Hogeweg, P.; Takeuchi, N. Multilevel Selection in Models of Prebiotic Evolution: Compartments and Spatial Self-organization. Orig. Life Evol. Biosph. 2003, 33, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. Unnatural Selection in Chemical Systems. Acc. Chem. Res. 1995, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligomer | m/z (+Na) | Normalized Abundance in 8D Sample 2 | Normalized Abundance in 8W Sample 2 |
|---|---|---|---|
| G2C2 1 | 537.2 | 1 | 1 |
| G3C2 | 611.3 | 2.50 | 1.87 |
| G4C2 * | 667.3 | 0.71 | 0.27 |
| G4C2 | 685.3 | 2.93 | 1.88 |
| G5C2 | 759.4 | 1.78 | 0.94 |
| G4C3 | 859.4 | 1.22 | 1.14 |
| G5C3 * | 915.4 | 0.96 | 0.41 |
| G5C3 | 933.4 | 2.18 | 1.21 |
| G6C3 * | 989.4 | 0.98 | 0.32 |
| G6C3 | 1007.4 | 1.63 | 0.85 |
| G7C3 | 1081.5 | 0.82 | 0.30 |
| G5C4 | 1107.5 | 0.47 | 0.37 |
| G6C4 | 1181.5 | 0.82 | 0.31 |
| G7C4 * | 1237.5 | 0.54 | 0.14 |
| G7C4 | 1255.6 | 1.01 | 0.52 |
| G8C4 | 1329.6 | 0.67 | 0.29 |
| Series/Cycles | DRY | D | W | Initial | WET |
|---|---|---|---|---|---|
| 0 | 4H:9.93H (1C:2.0G) | ||||
| 1 | 4H 1:9.89H (1C:2.0G) | 4H:9.89H (1C:2.0G) | 4H:9.81H (1C:2.0G) | ||
| 2 | 4H:10.14H (1C:2.0G) | 4H:9.94H (1C:2.0G) | 4H:9.92H (1C:2.0G) | ||
| 4 | 4H:14.03H (1C:2.8G) | 4H:10.20H (1C:2.0G) | 4H:10.28H (1C:2.1G) | ||
| 8 | 4H:11.99H (1C:2.4G) | 4H:10.37H (1C:2.1G) | 4H:9.74H (1C:1.9G) |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamajanov, I. Wet-Dry Cycling Delays the Gelation of Hyperbranched Polyesters: Implications to the Origin of Life. Life 2019, 9, 56. https://doi.org/10.3390/life9030056
Mamajanov I. Wet-Dry Cycling Delays the Gelation of Hyperbranched Polyesters: Implications to the Origin of Life. Life. 2019; 9(3):56. https://doi.org/10.3390/life9030056
Chicago/Turabian StyleMamajanov, Irena. 2019. "Wet-Dry Cycling Delays the Gelation of Hyperbranched Polyesters: Implications to the Origin of Life" Life 9, no. 3: 56. https://doi.org/10.3390/life9030056
APA StyleMamajanov, I. (2019). Wet-Dry Cycling Delays the Gelation of Hyperbranched Polyesters: Implications to the Origin of Life. Life, 9(3), 56. https://doi.org/10.3390/life9030056