Impact of Rearing Conditions on the Ambrosia Beetle’s Microbiome

 ,
,  ,
, 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Beetle Collections
2.2. Rearing Conditions
2.3. DNA Library Construction
2.4. Sequence Processing and OTUs Identification
2.5. Statistical Analyses of Identified OTUs
2.6. Metabolic Potential of the Fungal Communities
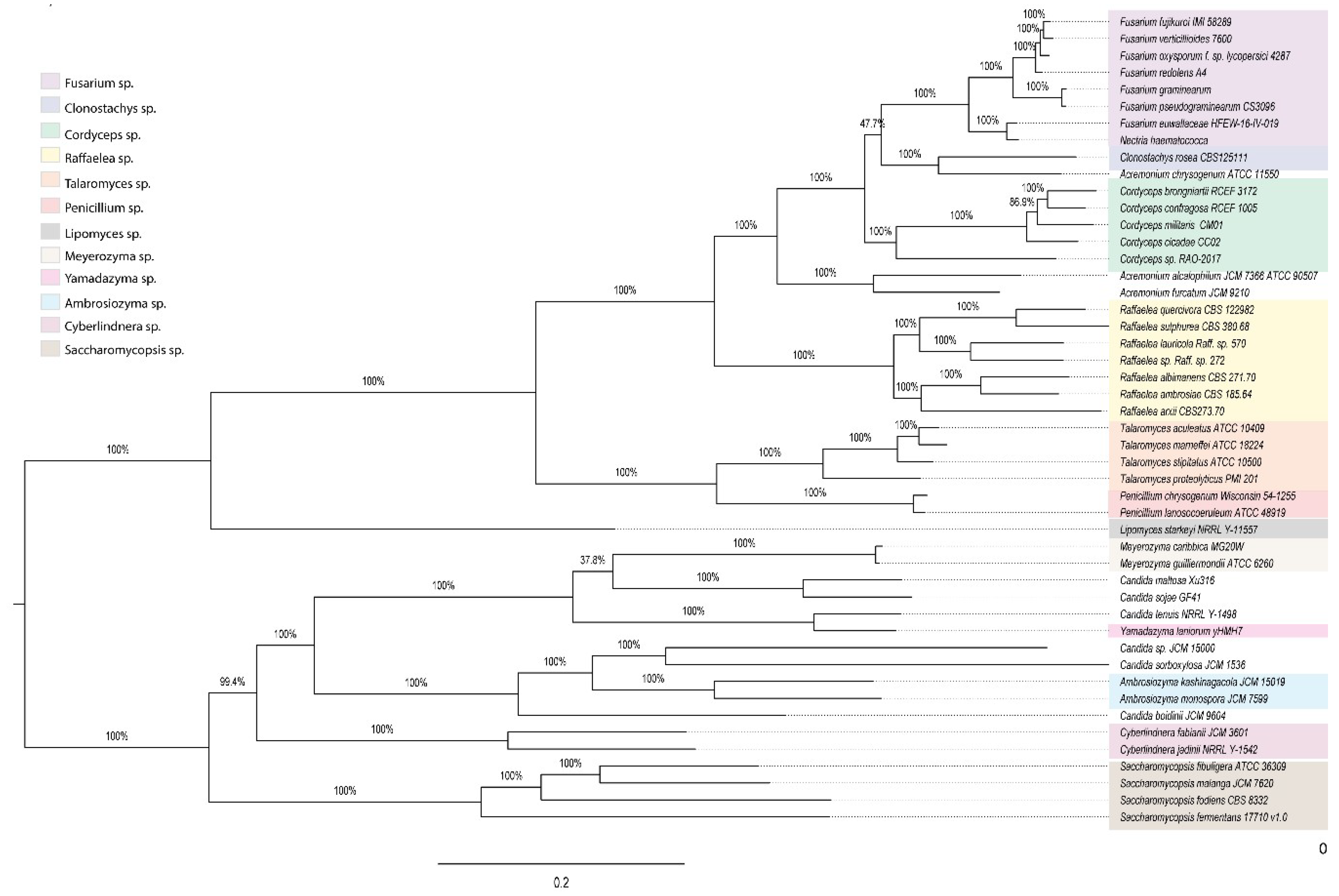
2.7. 18S rDNA Gene Phylogenetic Reconstruction
3. Results
3.1. Alpha Diversity Analysis of Bacterial and Fungal Microbiome
3.2. Core Microbiome of Beetles Reared under Laboratory and Wild Conditions
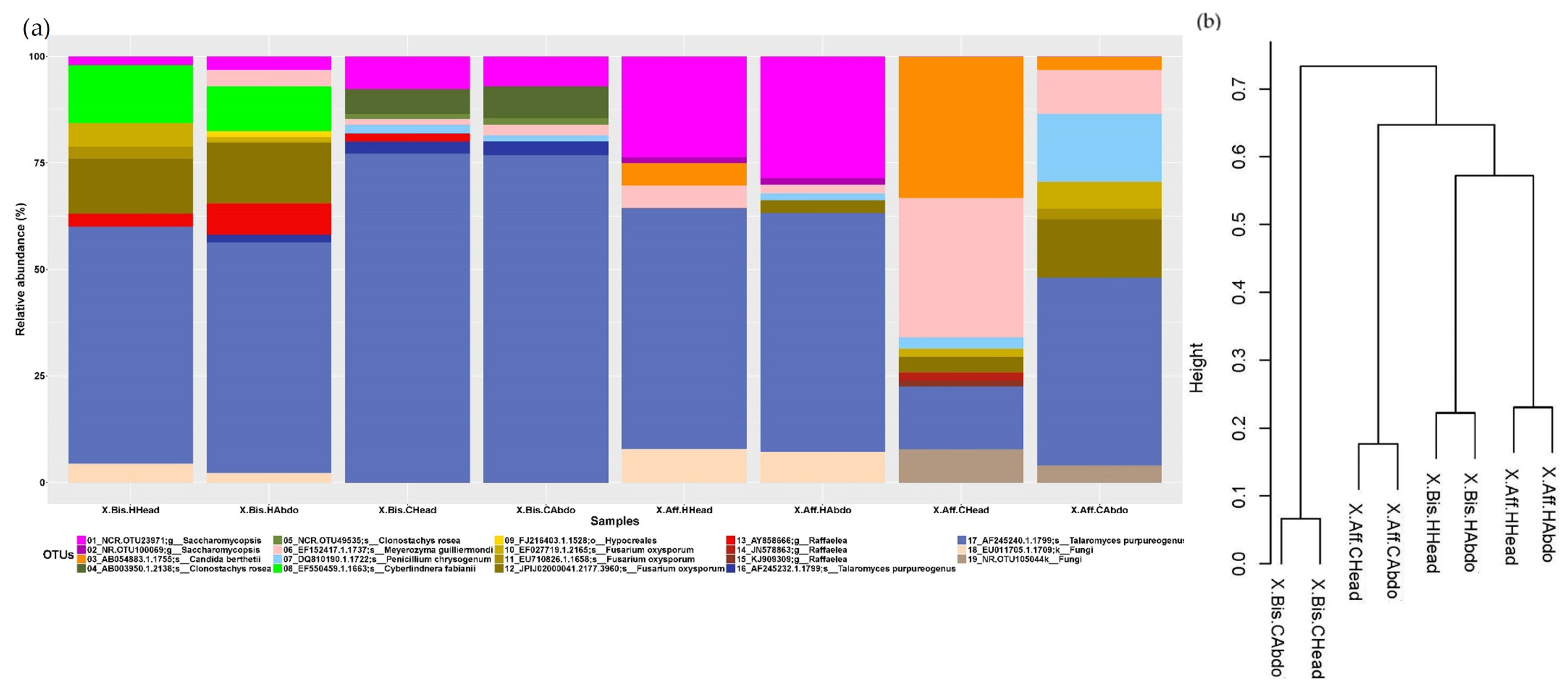
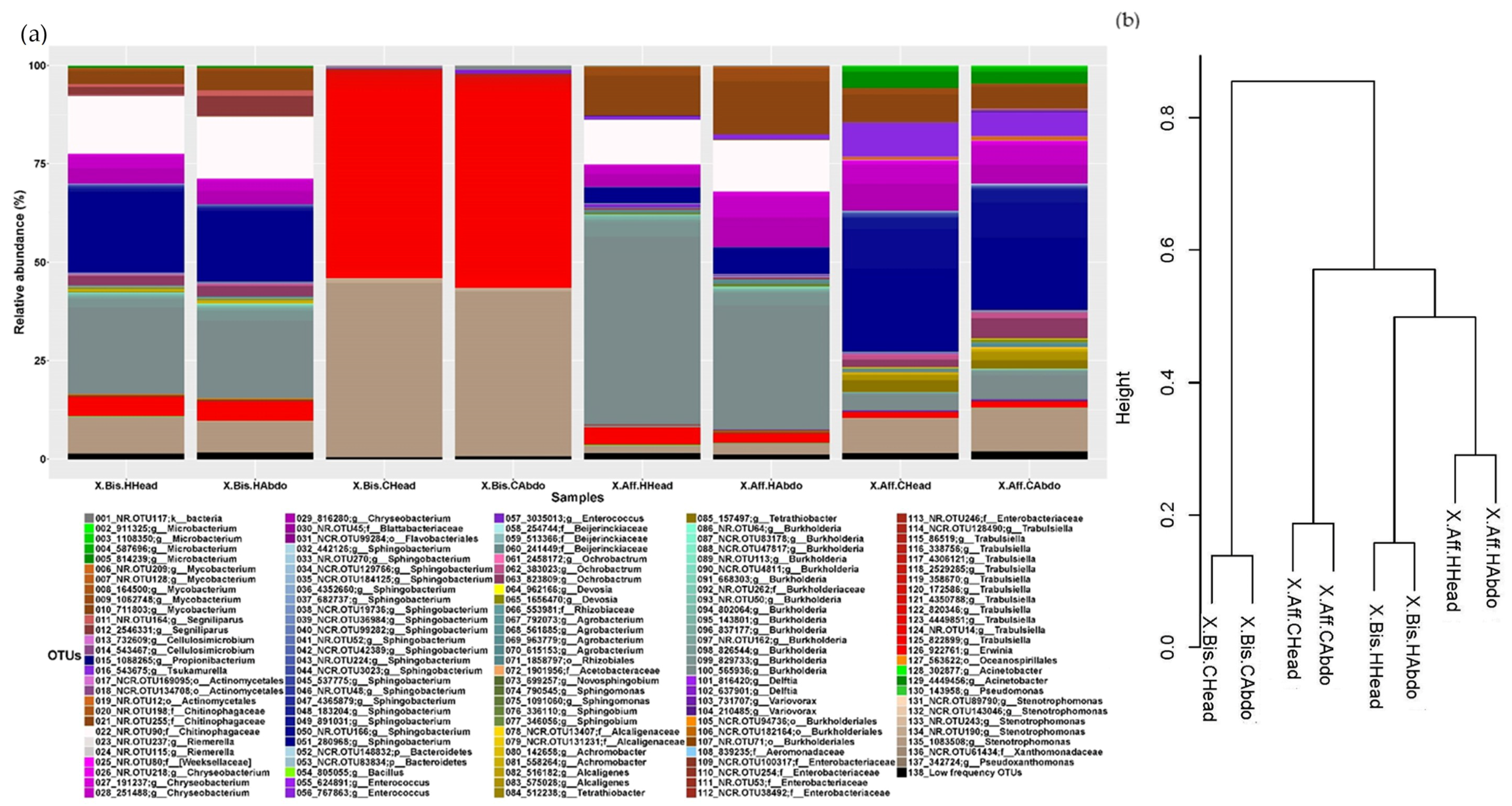
3.3. Microbiome Structure of Beetles Reared under Laboratory Conditions
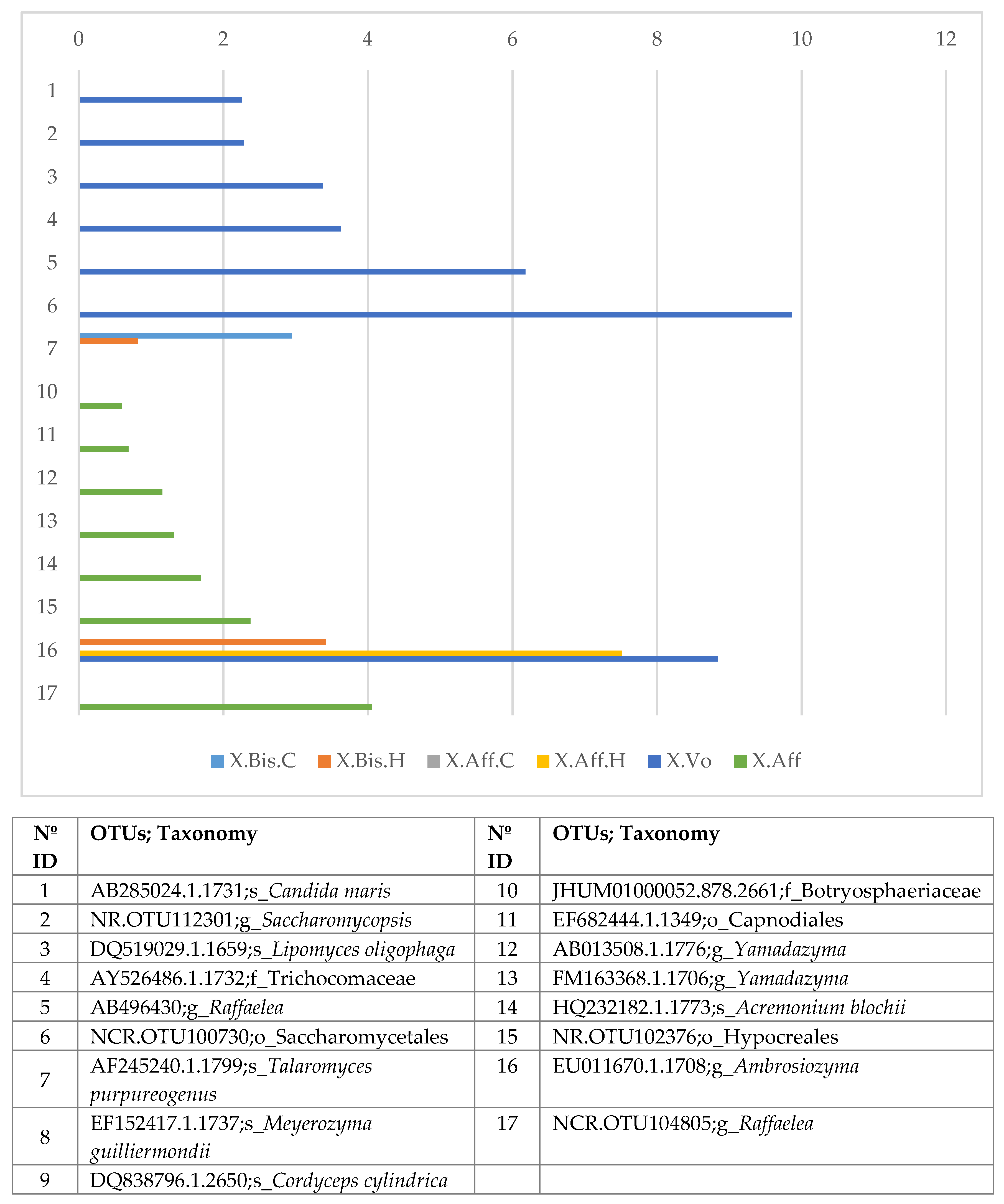
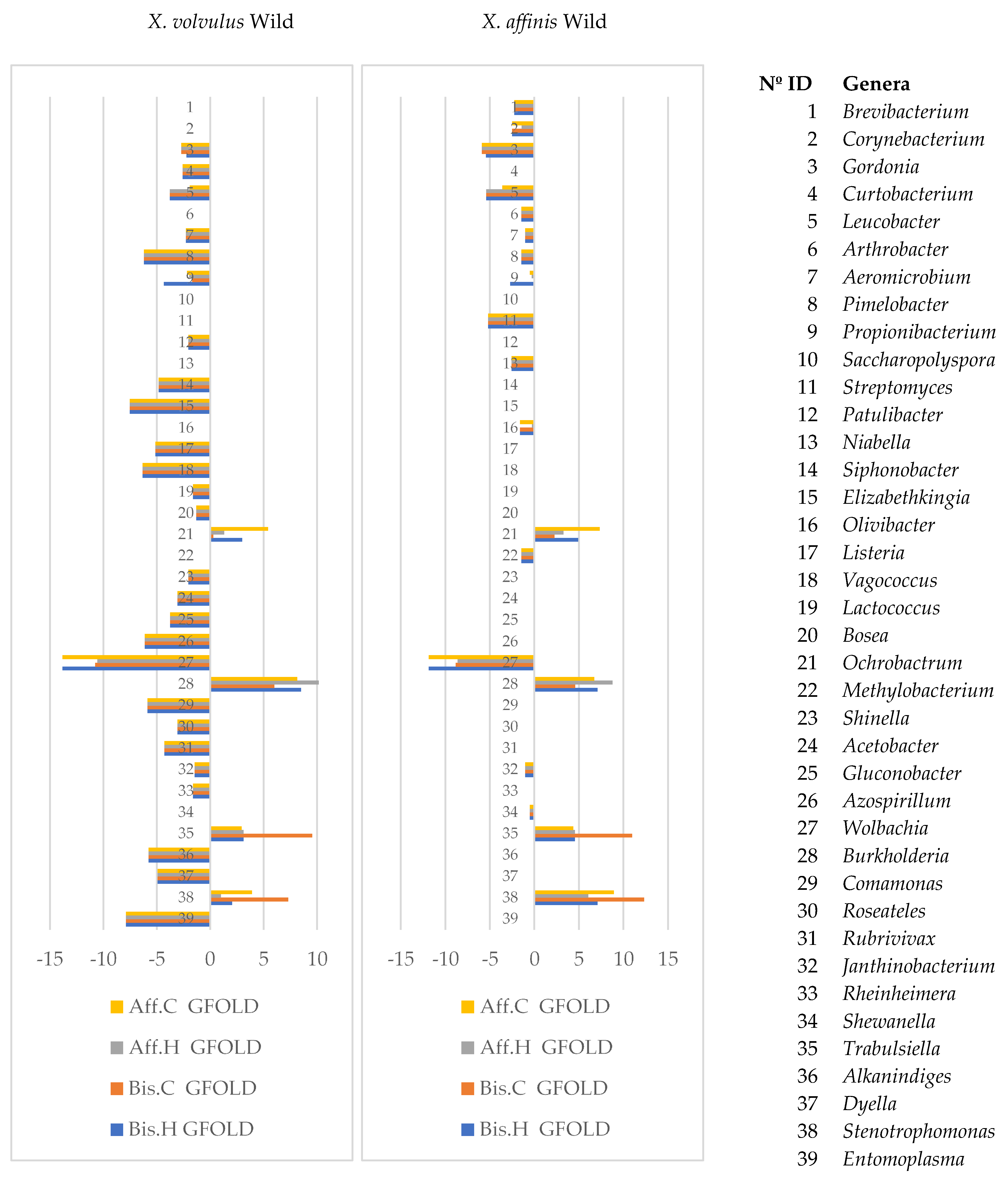
3.4. Microbiome Comparisons of Beetles Reared under Laboratory and Wild Conditions
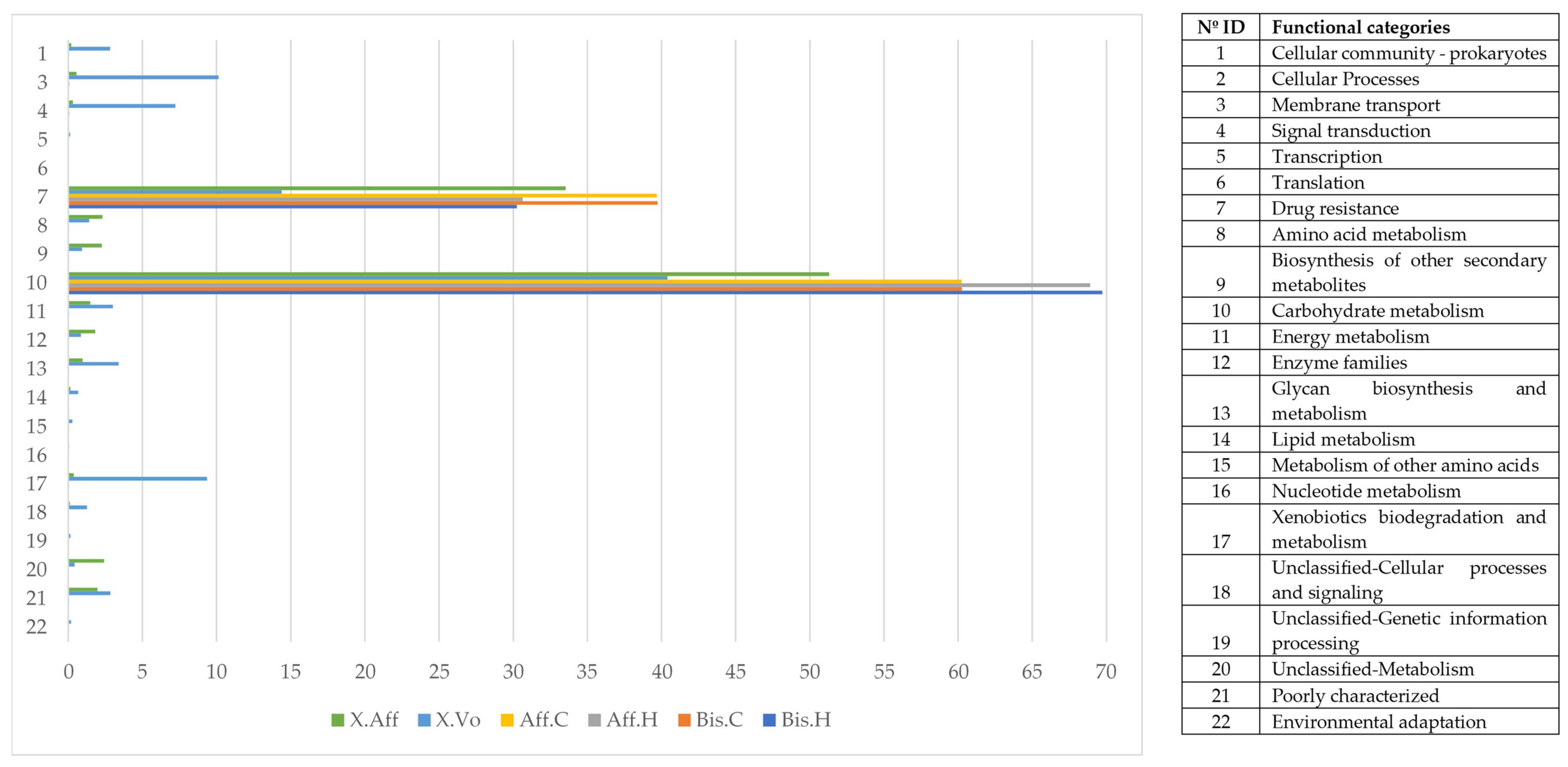
3.5. Functional Metabolic Prediction of Fungal Genera
3.6. Bacterial Functional Categories between Laboratory-Reared Beetles
3.7. Bacterial Functional Categories in Wild- Versus Laboratory-Reared Beetles
4. Discussion
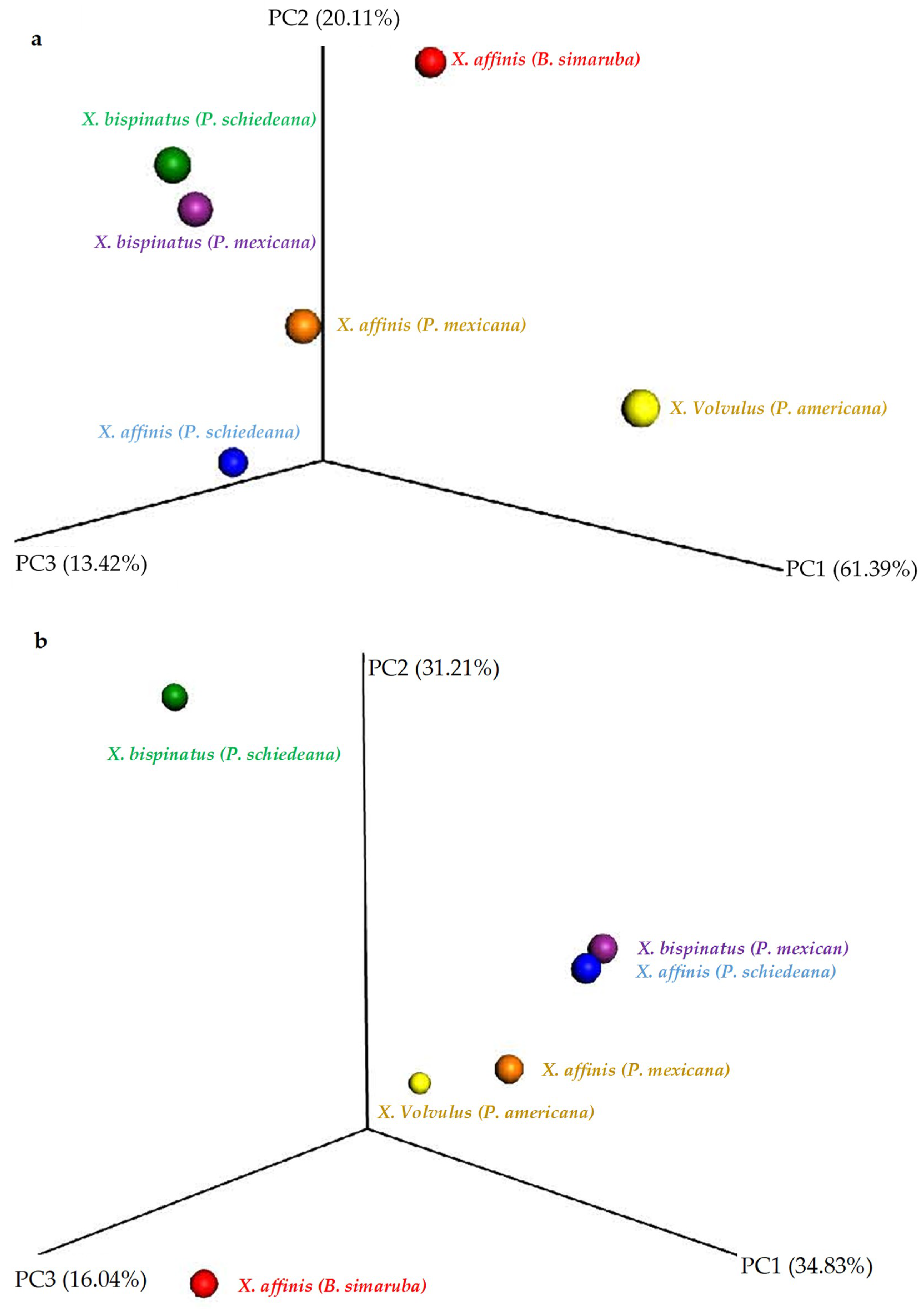
- (a)
- Head vs. abdomen of the same species reared on the same medium. Although the microbiomes in the head and abdomen were similar, the relative frequency of some OTUs, bacterial genera, and bacterial metabolic functions varied among samples. Changes in the microbiome structure depended on the rearing medium and the beetle species. In both cases, the differences in fungal and bacterial microbiome could be explained by the environmental conditions. The fungal and bacterial microbiomes in the head may be exposed to different pH, oxygen levels, and nutrient availability compared to the abdomen. Moreover, the role of the microbiome varies in the different parts of the beetle body; the head encloses the mycangia, which act as a receptacle to preserve microorganisms while the abdomen (gut) contains most of the beetle’s digestive system. The principal functions of the bacterial microbiome in the head were found to be related to communication between microbiomes: “Cellular Processes”, “Cellular community—prokaryotes”, “Cell motility”, “Signal transduction”, “Membrane transport”, “Transport and catabolism”, “Transcription”, “Drug resistance”, and “Biosynthesis of other secondary metabolites”, while in the abdomen the functions were related to nutrition: “Cell growth and death”, “Folding, sorting and degradation”, “Amino acid metabolism”, “Infectious diseases”, “Enzyme families”, “Energy metabolism”, “Carbohydrate metabolism”, “Xenobiotics biodegradation and metabolism”, “Nucleotide metabolism”, “Metabolism of terpenoids and polyketides”, “Metabolism of other amino acids”, “Metabolism of cofactors and vitamins”, and “Lipid metabolism”. These results are consistent with the functions of the head and abdomen in beetles. Surprisingly, the study of the functional abilities of the fungal genomes did not differ greatly between samples.
- (b)
- Same species reared on different media. The microbiome structure varied with the media and the beetle species, increasing the abundance of the bacterial genera Sphingobium, Burkholderia, Acinetobacter, Pseudomonas, and Mycobacterium in P. schiedeana compared to the beetles reared on P. mexicana. The metabolic categories “Transcription” and “Xenobiotics biodegradation and metabolism” increased in P. mexicana rearing medium, the fungal microbiome metabolic capabilities remained unchanged. The genera Acinetobacter and Pseudomonas were linked with phenolic glycoside metabolism in gypsy moth [17] and terpenes metabolism in Dendroctonus ponderosa [57]. Phenolic and terpenoids compounds they are part of the plant defenses and has been describe the role of the bark beetle’s microbiome in the detoxification of these molecules [17]. The increment of these bacterial genera in P. schiedeana could be explain for a modification of the levels of these components in the rearing medium. This hypothesis should be verified.
- (c)
- Wild vs. laboratory rearing conditions. All the laboratory samples exhibited large abundance on T. purpureogenus and M. guilliermondii. Regarding the bacterial microbiome, 62 OTUs were more abundant in the wild than in lab-reared samples. The genera Gordonia, Leucobacter, Aeromicrobium, Pimelobacter, Propionibacterium, Wolbachia, and Janthinobacterium were more abundant in wild than in laboratory samples, while Ochrobactrum, Burkholderia, Trabulsiella, and Stenotrophomonas were more abundant in laboratory than in wild beetles. All these genera have been found in other insect microbiomes [3,13,17,58]. The fungal metabolic categories did not differ between the microbiomes associated to the different rearing conditions. The metabolic categories with greater abundance in wild than laboratory conditions were related to: “Basic cellular functions”: Transcription and translation, cellular processes, and signaling. “Metabolism”: Obtaining energy and production of basic components. “Defense”: Metabolism of secondary metabolites, Degradation of xenobiotics, Signal transduction (Bacterial toxins and Two-component system). “Communication”: Cell communication, Membrane, and Plant-pathogen interaction.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Dimijian, G.G. Evolving together: The biology of symbiosis, part 1. Proc. (Bayl. Univ. Med. Cent.) 2000, 13, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Aylward, F.O.; Suen, G.; Biedermann, P.H.W.; Adams, A.S.; Scott, J.J.; Malfatti, S.A.; Glavina del Rio, T.; Tringe, S.G.; Poulsen, M.; Raffa, K.F.; et al. Convergent bacterial microbiotas in the fungal agricultural systems of insects. MBio 2014, 5, e02077. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.G.; Gerardo, N. Fungus-farming insects: Multiple origins and diverse evolutionary histories. Proc. Natl. Acad. Sci. USA 2002, 99, 15247–15249. [Google Scholar] [CrossRef] [PubMed]
- Koch, F.H.; Smith, W.D. Spatio-temporal analysis of Xyleborus glabratus (Coleoptera: Curculionidae [corrected] Scolytinae) invasion in eastern U.S. forests. Environ. Entomol. 2008, 37, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Kostovcik, M.; Bateman, C.C.; Kolarik, M.; Stelinski, L.L.; Jordal, B.H.; Hulcr, J. The ambrosia symbiosis is specific in some species and promiscuous in others: Evidence from community pyrosequencing. ISME J. 2015, 9, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Batra, L.R. Ecology of Ambrosia Fungi and Their Dissemination by Beetles. Trans. Kans. Acad. Sci. 1963, 66, 213–236. [Google Scholar] [CrossRef]
- Hulcr, J.; Stelinski, L.L. The Ambrosia Symbiosis: From Evolutionary Ecology to Practical Management. Annu. Rev. Entomol. 2017, 62, 285–303. [Google Scholar] [CrossRef]
- Six, D.L. Ecological and Evolutionary Determinants of Bark Beetle -Fungus Symbioses. Insects 2012, 3, 339–366. [Google Scholar] [CrossRef]
- Kirkendall, L.R.; Biedermann, P.H.W.; Jordal, B.H. Chapter 3—Evolution and Diversity of Bark and Ambrosia Beetles. In Bark Beetles; Vega, F.E., Hofstetter, R.W., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 85–156. ISBN 978-0-12-417156-5. [Google Scholar]
- Wood, S.L. The bark and ambrosia beetles of North and Central America (Coleoptera: Scolytidae): A taxonomic monograph [North America]; Brigham Young University: Provo, UT, USA, 1982. [Google Scholar]
- Hulcr, J.; Mogia, M.; Isua, B.; Novotny, V. Host specificity of ambrosia and bark beetles (Col., Curculionidae: Scolytinae and Platypodinae) in a New Guinea rainforest. Ecol. Entomol. 2007, 32, 762–772. [Google Scholar] [CrossRef]
- Hulcr, J.; Rountree, N.R.; Diamond, S.E.; Stelinski, L.L.; Fierer, N.; Dunn, R.R. Mycangia of ambrosia beetles host communities of bacteria. Microb. Ecol. 2012, 64, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.S.; Ploetz, R.C.; Dreaden, T.J.; Kendra, P.E.; Montgomery, W.S. Geographic variation in mycangial communities of Xyleborus glabratus. Mycologia 2016, 108, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Choi, M.-Y.; Kim, J.-W.; Lee, S.A.; Ahn, J.-H.; Song, J.; Kim, S.-H.; Weon, H.-Y. Effects of diet type, developmental stage, and gut compartment in the gut bacterial communities of two Cerambycidae species (Coleoptera). J. Microbiol. 2017, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.J.; Hanshew, A.S.; Raffa, K.F. Contributions by Host Trees and Insect Activity to Bacterial Communities in Dendroctonus valens (Coleoptera: Curculionidae) Galleries, and Their High Overlap With Other Microbial Assemblages of Bark Beetles. Environ. Entomol. 2016, 45, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Eskalen, A.; Stouthamer, R.; Lynch, S.C.; Rugman-Jones, P.F.; Twizeyimana, M.; Gonzalez, A.; Thibault, T. Host Range of Fusarium Dieback and Its Ambrosia Beetle (Coleoptera: Scolytinae) Vector in Southern California. Plant Dis. 2013, 97, 938–951. [Google Scholar] [CrossRef]
- Brar, G.S.; Capinera, J.L.; Kendra, P.E.; McLean, S.; Peña, J.E. Life Cycle, Development, and Culture of Xyleborus glabratus (Coleoptera: Curculionidae: Scolytinae). Fla. Entomol. 2013, 96, 1158–1167. [Google Scholar] [CrossRef]
- Tremmel, M.; Müller, C. Insect personality depends on environmental conditions. Behav. Ecol. 2012, 24, 386–392. [Google Scholar] [CrossRef]
- Barrera, J.F.; Villacorta, A.; Herrera, J. Seasonal fluctuation of captures of the “coffee berry borer” (Hypothenemus hampei) with methanol—Ethanol traps and implications on sample size. Entomol. Mex 2004, 3, 540–544. [Google Scholar]
- Carrillo, D.; Amalin, D.; Hosein, F.; Roda, A.; Duncan, R.E.; Peña, J.E. Host plant range of Raoiella indica (Acari: Tenuipalpidae) in areas of invasion of the New World. Exp. Appl. Acarol. 2012, 57, 271–289. [Google Scholar] [CrossRef]
- Pérez-De La Cruz, M.; Equihua-Martínez, A.; Romero-Nápoles, J.; Sánchez-Soto, S.; García-López, E. Diversidad, fluctuación poblacional y plantas huésped de escolitinos (Coleoptera: Curculionidae) asociados con el agroecosistema cacao en Tabasco, México. Rev. Mex. Biodivers. 2009, 80, 779–791. [Google Scholar]
- Wood, S.L. A reclassification of the genera of Scolytidae (Coleoptera). Great Basin Nat. Mem. 1986, 10, 2. [Google Scholar]
- Hanula, J.L.; Mayfield, A.E.; Fraedrich, S.W.; Rabaglia, R.J. Biology and host associations of redbay ambrosia beetle (Coleoptera: Curculionidae: Scolytinae), exotic vector of laurel wilt killing redbay trees in the southeastern United States. J. Econ. Entomol. 2008, 101, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Menocal, O.; Cruz, L.F.; Kendra, P.E.; Crane, J.H.; Ploetz, R.C.; Carrillo, D. Rearing Xyleborus volvulus (Coleoptera: Curculionidae) on Media Containing Sawdust from Avocado or Silkbay, With or Without Raffaelea lauricola (Ophiostomatales: Ophiostomataceae). Environ. Entomol. 2017, 46, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Moya, A.; Ayala, F.J. Evolution of mitochondrial DNA in Drosophila subobscura. Proc. Natl. Acad. Sci. USA 1986, 83, 8649–8653. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. Gigascience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes, R package version 1.0-2; GitHub, Inc.: San Francisco, CA, USA, 2007. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013.
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley Liu, X.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Lomsadze, A.; Ter-Hovhannisyan, V.; Chernoff, Y.O.; Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez Alvarado, A.; Yandell, M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. UniProtKB/Swiss-Prot. Methods Mol. Biol. 2007, 406, 89–112. [Google Scholar] [PubMed]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Obadia, B.; Keebaugh, E.S.; Yamada, R.; Ludington, W.B.; Ja, W.W. Diet influences host-microbiota associations in Drosophila. Proc. Natl. Acad. Sci. USA 2018, 115, E4547–E4548. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.K.; Keefover-Ring, K.; Mapes, A.C.; Adams, A.S.; Bohlmann, J.; Raffa, K.F. Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J. Chem. Ecol. 2013, 39, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Hernández-García, J.A.; Gonzalez-Escobedo, R.; Briones-Roblero, C.I.; Cano-Ramírez, C.; Rivera-Orduña, F.N.; Zúñiga, G. Gut Bacterial Communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): A Metagenomic Analysis across Different Geographical Locations in Mexico. Int. J. Mol. Sci. 2018, 19, 2578. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Turkarslan, S.; Beer, K.D.; Lo, F.Y.; Baliga, N.S. Adaptation of cells to new environments. Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 544–561. [Google Scholar] [CrossRef]
- Bobay, L.-M.; Ochman, H. Factors driving effective population size and pan-genome evolution in bacteria. BMC Evol. Biol. 2018, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Soen, Y. Environmental disruption of host–microbe co-adaptation as a potential driving force in evolution. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.L.; Ling, H.; Lee, Y.S.; Chang, M.W. Microbiome engineering: Current applications and its future. Biotechnol. J. 2017, 12, 1600099. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Samples | Fungal Samples | ||||||
|---|---|---|---|---|---|---|---|
| ID Samples | Specie Name | Observed OTUs | Shannon Index | Simpson Index | Observed OTUs | Shannon Index | Simpson Index |
| X.Aff.CAbdo | X. affinis | 199 | 4.596 | 0.925 | 8 | 2.405 | 0.744 |
| X.Aff.CHead | X. affinis | 180 | 4.347 | 0.912 | 9 | 2.368 | 0.752 |
| X.Aff.HAbdo | X. affinis | 158 | 3.713 | 0.850 | 7 | 1.714 | 0.597 |
| X.Aff.HHead | X. affinis | 169 | 3.231 | 0.743 | 6 | 1.783 | 0.614 |
| X.Bis.CAbdo | X. bispinatus | 73 | 1.813 | 0.597 | 7 | 1.314 | 0.397 |
| X.Bis.CHead | X. bispinatus | 64 | 1.728 | 0.591 | 8 | 1.343 | 0.393 |
| X.Bis.HAbdo | X. bispinatus | 178 | 4.034 | 0.888 | 10 | 2.232 | 0.667 |
| X.Bis.HHead | X. bispinatus | 178 | 3.904 | 0.874 | 8 | 2.085 | 0.648 |
| X.Aff | X. affinis | 312 | 3.447 | 0.744 | 15 | 2.438 | 0.669 |
| X.Vo | X. volvulus | 192 | 3.631 | 0.758 | 9 | 1.984 | 0.588 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarra-Juarez, L.A.; Desgarennes, D.; Vázquez-Rosas-Landa, M.; Villafan, E.; Alonso-Sánchez, A.; Ferrera-Rodríguez, O.; Moya, A.; Carrillo, D.; Cruz, L.; Carrión, G.; et al. Impact of Rearing Conditions on the Ambrosia Beetle’s Microbiome. Life 2018, 8, 63. https://doi.org/10.3390/life8040063
Ibarra-Juarez LA, Desgarennes D, Vázquez-Rosas-Landa M, Villafan E, Alonso-Sánchez A, Ferrera-Rodríguez O, Moya A, Carrillo D, Cruz L, Carrión G, et al. Impact of Rearing Conditions on the Ambrosia Beetle’s Microbiome. Life. 2018; 8(4):63. https://doi.org/10.3390/life8040063
Chicago/Turabian StyleIbarra-Juarez, Luis Arturo, Damaris Desgarennes, Mirna Vázquez-Rosas-Landa, Emanuel Villafan, Alexandro Alonso-Sánchez, Ofelia Ferrera-Rodríguez, Andrés Moya, Daniel Carrillo, Luisa Cruz, Gloria Carrión, and et al. 2018. "Impact of Rearing Conditions on the Ambrosia Beetle’s Microbiome" Life 8, no. 4: 63. https://doi.org/10.3390/life8040063
APA StyleIbarra-Juarez, L. A., Desgarennes, D., Vázquez-Rosas-Landa, M., Villafan, E., Alonso-Sánchez, A., Ferrera-Rodríguez, O., Moya, A., Carrillo, D., Cruz, L., Carrión, G., López-Buenfil, A., García-Avila, C., Ibarra-Laclette, E., & Lamelas, A. (2018). Impact of Rearing Conditions on the Ambrosia Beetle’s Microbiome. Life, 8(4), 63. https://doi.org/10.3390/life8040063