G-Protein Coupled Receptor Protein Synthesis on a Lipid Bilayer Using a Reconstituted Cell-Free Protein Synthesis System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of DNA Constructs
2.3. Membrane Scaffold Protein (MSP) Expression
2.4. Nanodisc (ND) Preparation
2.5. PURE Synthesis of Membrane Proteins
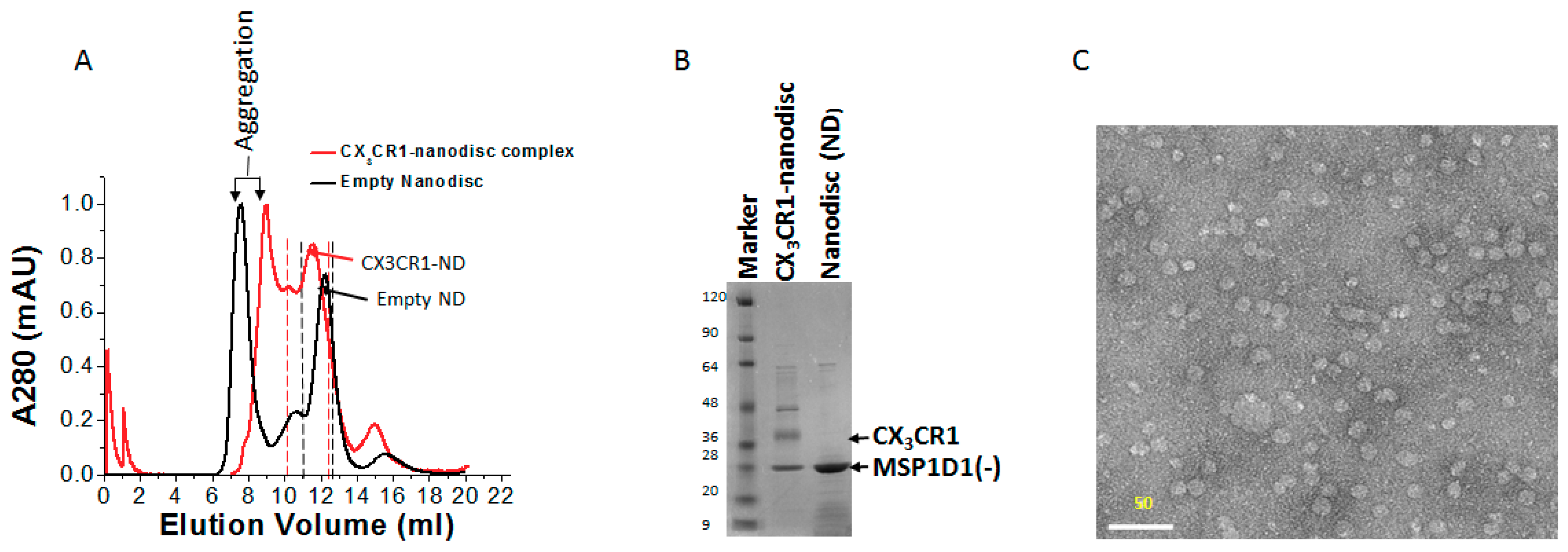
2.6. Purification of Membrane Protein-Nanodisc Complex
2.7. Circular Dichroism (CD) Spectroscopy
2.8. Preparation of Membrane Proteins for CD Spectroscopy by Micelle Method
2.9. Electron Microscopy of CX3CR1-Nanodisc Complexes
2.10. Giant Unilamellar Vesicle (GUV) Preparation
2.11. Determination of Binding Constants by the Surface Plasmon Resonance (SPR)
2.12. Confocal Microscopy
3. Results
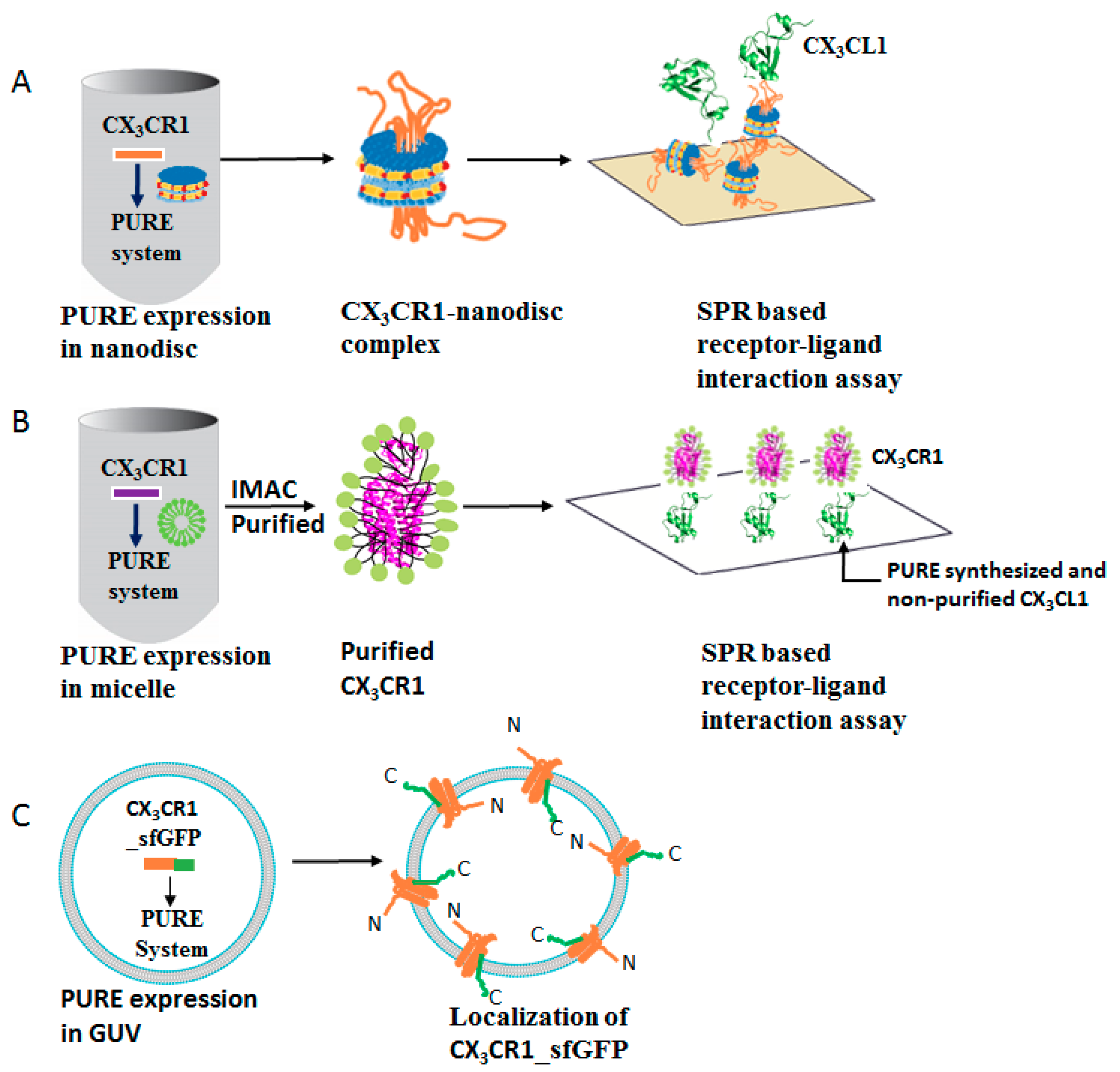
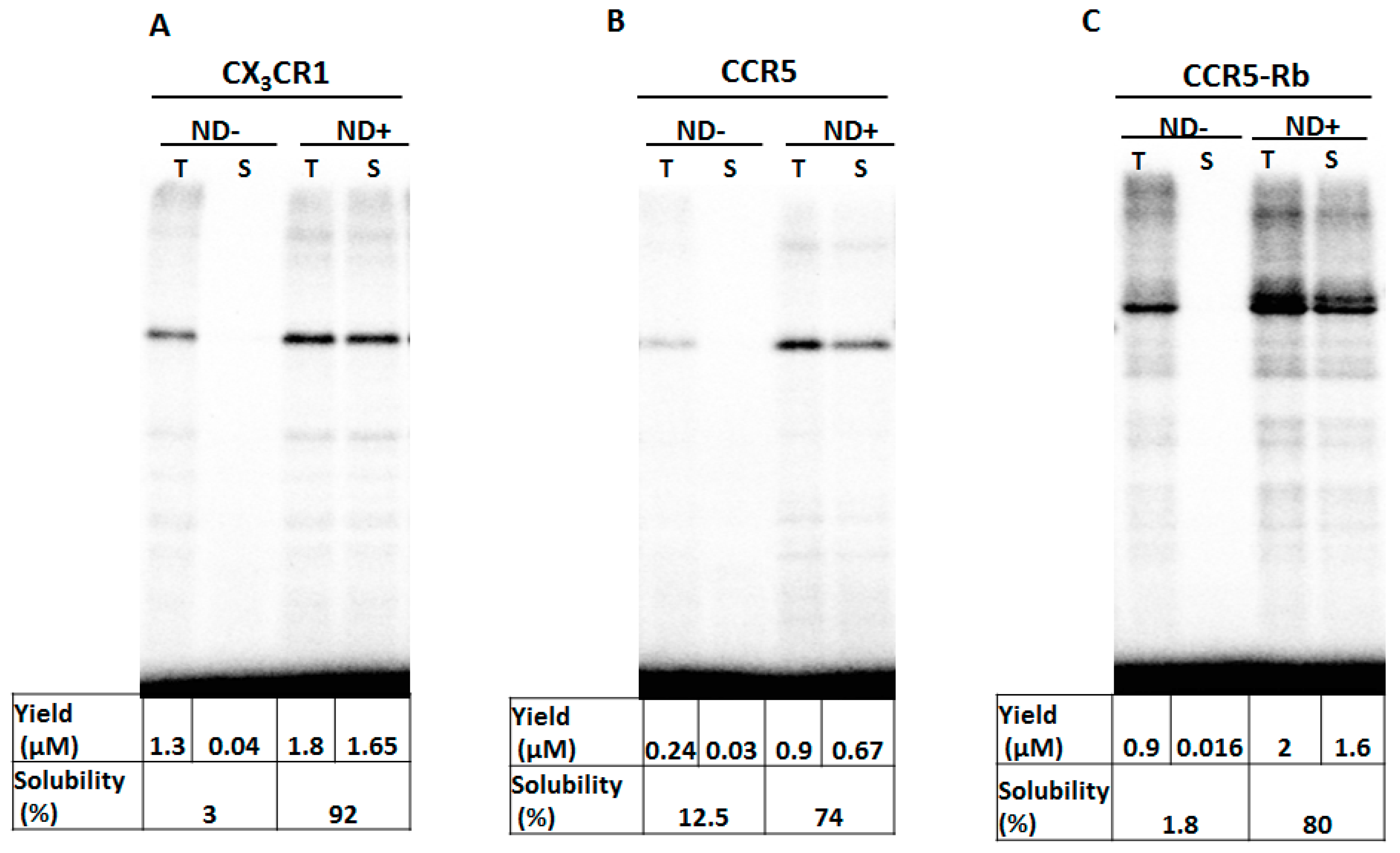
3.1. Membrane Protein Synthesis in Lipid Nanodisc
3.2. Membrane Protein Synthesis in Micelle
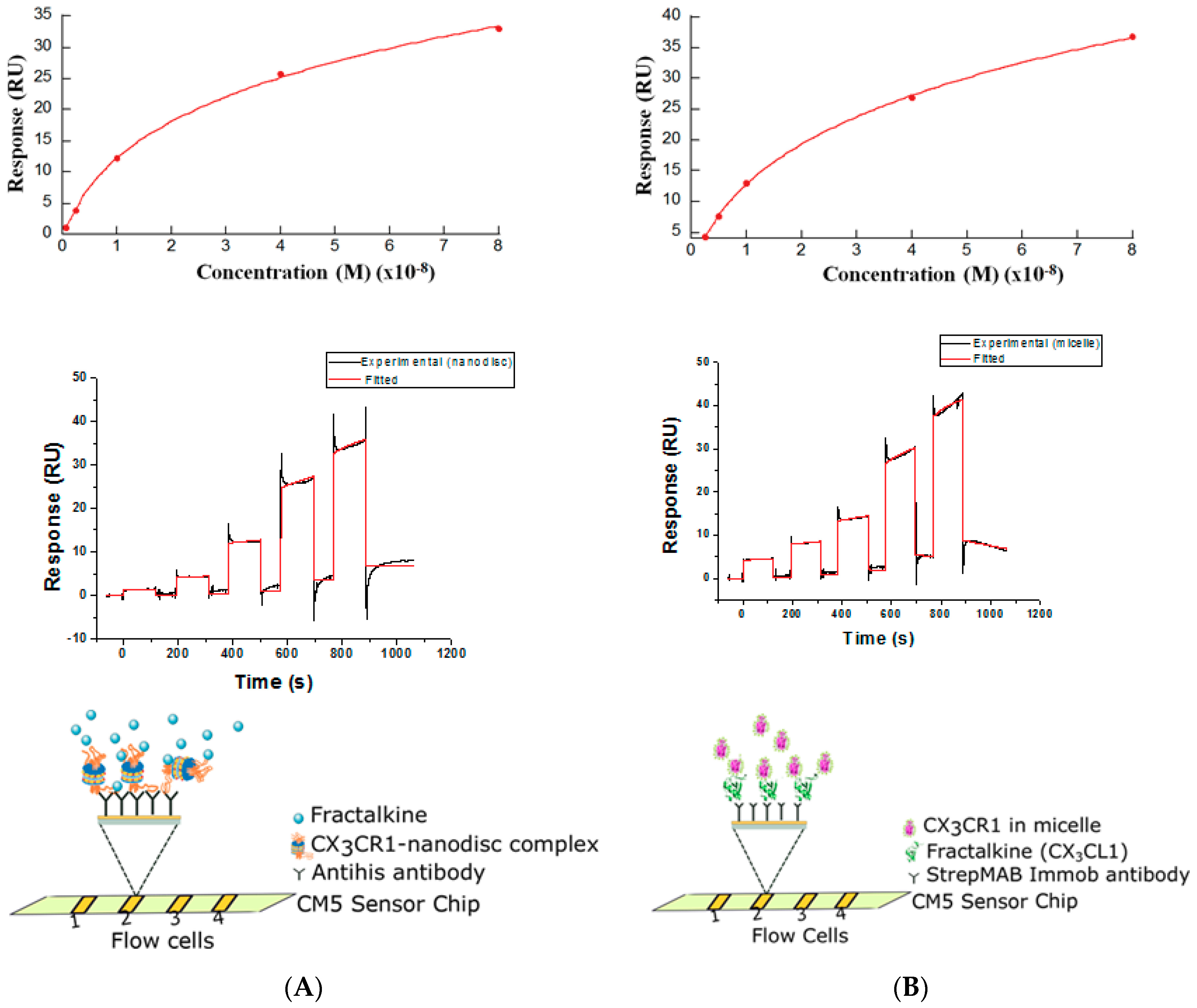
3.3. Membrane Protein Synthesis in Nanodisc and Micelle
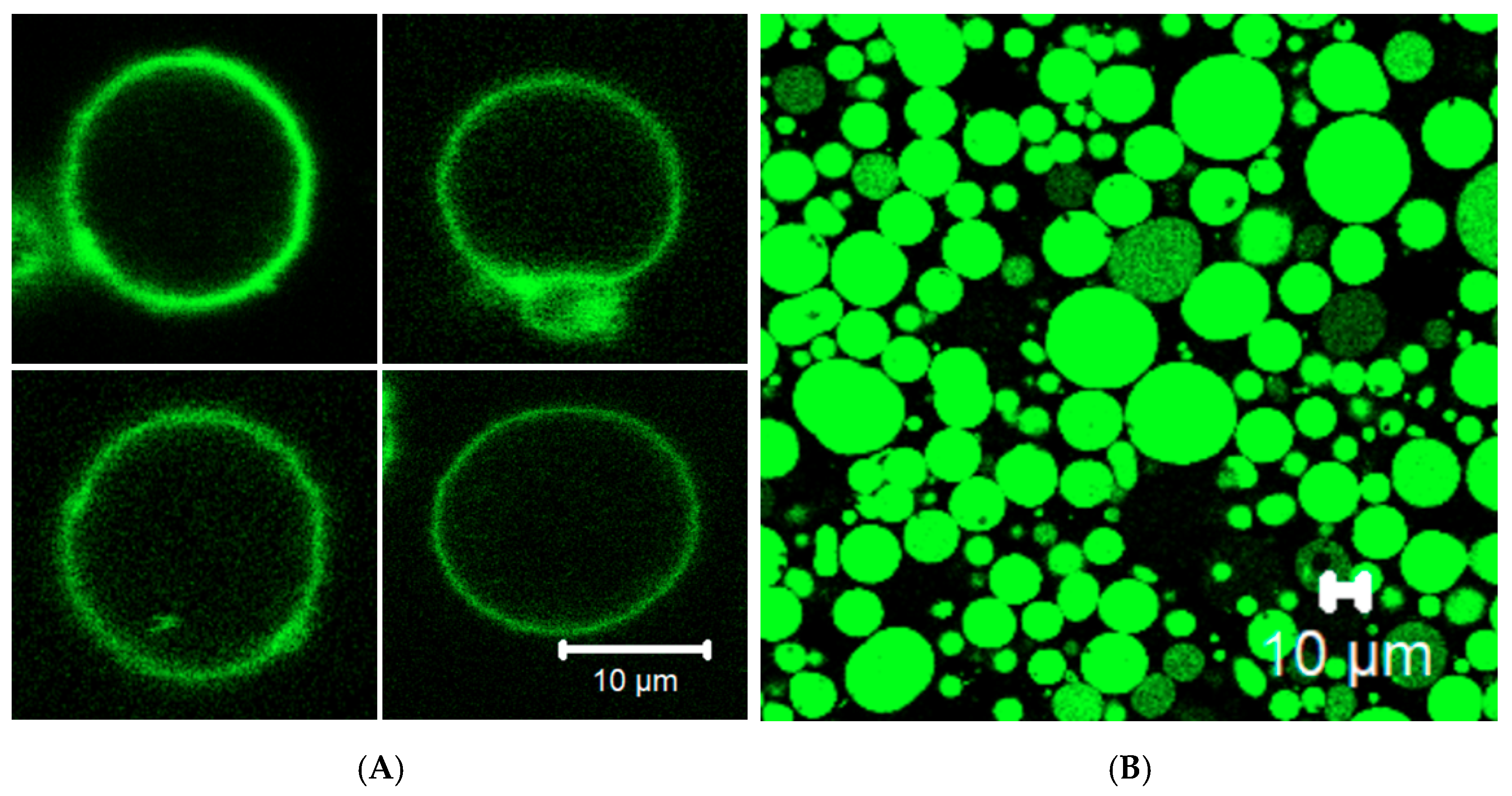
3.4. Membrane Protein Synthesis in GUV
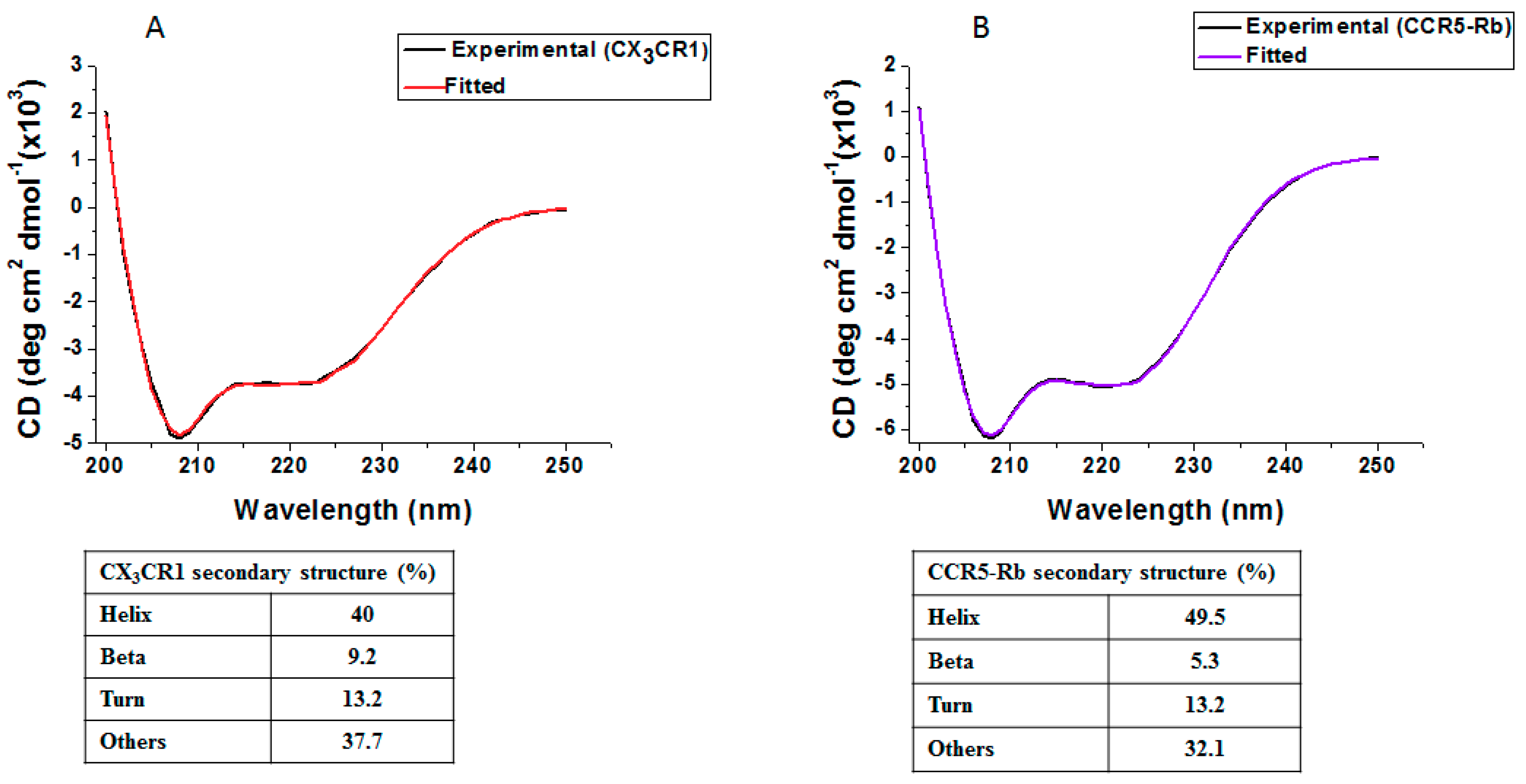
3.5. Membrane Protein Synthesis in Nanodisc for Structural Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shen, H.H.; Lithgow, T.; Martin, L. Reconstitution of Membrane Proteins into Model Membranes: Seeking Better Ways to Retain Protein Activities. Int. J. Mol. Sci. 2013, 14, 1589–1607. [Google Scholar] [CrossRef] [PubMed]
- Sachse, R.; Dondapati, S.K.; Fenz, S.F.; Schmidt, T.; Kubick, S. Membrane protein synthesis in cell-free systems: From bio-mimetic systems to bio-membranes. FEBS Lett. 2014, 588, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wüthrich, K. The GPCR Network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Noireaux, V.; Libchaber, A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 17669–17674. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Matsuura, T.; Sunami, T.; Nishikawa, T.; Kazuta, Y.; Yomo, T. Liposome display for in vitro selection and evolution of membrane proteins. Nat. Protoc. 2014, 9, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Iwamoto, M.; Kato, A.; Yoshikawa, K.; Oiki, S. Oriented reconstitution of a membrane protein in a giant unilamellar vesicle: Experimental verification with the potassium channel KcsA. J. Am. Chem. Soc. 2011, 133, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Tabuchi, M.; Toyota, T.; Sakurai, T.; Hosoi, T.; Nomoto, T.; Nakatani, K.; Fujinami, M.; Kanzaki, R. Giant vesicles functionally expressing membrane receptors for an insect pheromone. Chem. Commun. (Camb.) 2014, 50, 2958–2961. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N.; Osaki, T.; Takeuchi, S. Membrane protein-based biosensors. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Wang, X.; Li, J.; Ren, H.; Huang, F. Folding of newly translated membrane protein CCR5 is assisted by the chaperonin GroEL-GroES. Sci. Rep. 2015, 5, 17037. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, T.; Shinya, N.; Ito, K.; Ishizuka-Katsura, Y.; Ohsawa, N.; Terada, T.; Hirata, K.; Kawano, Y.; Yamamoto, M.; Tomita, T.; et al. Cell-free methods to produce structurally intact mammalian membrane proteins. Sci. Rep. 2016, 6, 30442. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, H.; Kuruma, Y.; Ueda, T. In vitro synthesis of the, E. coli Sec translocon from DNA. Angew. Chem. Int. Ed. Engl. 2014, 53, 7535–7538. [Google Scholar] [CrossRef] [PubMed]
- Rues, R.B.; Dong, F.; Dötsch, V.; Bernhard, F. Systematic optimization of cell-free synthesized human endothelin B receptor folding. Methods 2018, 147, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Ghirlando, R.; Grisshammer, R. Biophysical characterization of membrane proteins in nanodiscs. Methods 2013, 59, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Yoshiura, C.; Kofuku, Y.; Ueda, T.; Mase, Y.; Yokogawa, M.; Osawa, M.; Terashima, Y.; Matsushima, K.; Shimada, I. NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers. J. Am. Chem. Soc. 2010, 32, 6768–6777. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Li, Y.; Yoon, S.H.; Ernst, R.K.; Walz, T.; Liao, M. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 2017, 549, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Rues, R.B.; Dötsch, V.; Bernhard, F. Co-translational formation and pharmacological characterization of beta1-adrenergic receptor/nanodisc complexes with different lipid environments. Biochim. Biophys. Acta 2016, 1858, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Klammt, C.; Srivastava, A.; Eifler, N.; Junge, F.; Beyermann, M.; Schwarz, D.; Michel, H.; Doetsch, V.; Bernhard, F. Functional analysis of cell-free-produced human endothelin B receptor reveals transmembrane segment 1 as an essential area for ET-1 binding and homodimer formation. FEBS J. 2007, 274, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Efremov, R.G.; Gatsogiannis, C.; Raunser, S. Lipid Nanodiscs as a Tool for High-Resolution Structure Determination of Membrane Proteins by Single-Particle Cryo-EM. Methods Enzymol. 2017, 594, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kuruma, Y.; Kanamori, T.; Ueda, T. The PURE system for protein production. Methods Mol. Biol. 2014, 1118, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.K.; Grinkova, Y.V.; Bayburt, T.H.; Denisov, I.G.; Zolnerciks, J.K.; Atkins, W.M.; Sligar, S.G. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Lazarides, A.A.; Sligar, S.G. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, R.; Liu, J.J.; Pauszek, R.F.; Millar, D.P. Fluorophore Labeling, Nanodisc Reconstitution and Single-molecule Observation of a G Protein-coupled Receptor. Bio-Protocol 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ueda, T. PURE technology. Methods Mol. Biol. 2010, 607, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Henrich, E.; Dötsch, V.; Bernhard, F. Screening for lipid requirements of membrane proteins by combining cell-free expression with nanodiscs. Methods Enzymol. 2015, 556, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.M.; Mizoue, L.S.; Handel, T.M.; Lubkowski, J. The Crystal Structure of the Chemokine Domain of Fractalkine Shows a Novel Quaternary Arrangement. J. Biol. Chem. 2000, 275, 23187–23193. [Google Scholar] [CrossRef] [PubMed]
- Shilling, P.J.; Bumbak, F.; Scott, D.J.; Bathgate, R.A.D.; Gooley, P.R. Characterisation of a cell-free synthesised G-protein coupled receptor. Sci. Rep. 2017, 7, 1094. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.L.; Steuerwald, D.; Kaiser, L.; Graveland-Bikker, J.; Vanberghem, M.; Berke, A.P.; Herlihy, K.; Pick, H.; Vogel, H.; Zhang, S. Large-scale production and study of a synthetic G protein-coupled receptor: Human olfactory receptor 17-4. Proc. Natl. Acad. Sci. USA 2009, 106, 11925–11930. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Yu, D.; Ge, B.; Cook, B.; Xu, Z.; Zhang, S. High-level production, solubilization and purification of synthetic human GPCR chemokine receptors CCR5, CCR3, CXCR4 and CX3CR1. PLoS ONE 2009, 4, e4509. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef] [PubMed]
- Wiktor, M.; Morin, S.; Sass, H.J.; Kebbel, F.; Grzesiek, S. Biophysical and structural investigation of bacterially expressed and engineered CCR5, a G protein-coupled receptor. J. Biomol. NMR 2013, 55, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Corin, K.; Baaske, P.; Ravel, D.B.; Song, J.; Brown, E.; Wang, X.; Geissler, S.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; et al. A robust and rapid method of producing soluble, stable, and functional G-protein coupled receptors. PLoS ONE 2011, 6, e23036. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Myburgh, R.; Krause, K.H. The chemokine receptor CCR5 in the central nervous system. Prog. Neurobiol. 2011, 93, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, S.; van Haastert, E.S.; Kuipers, H.F.; van den Elsen, P.J.; De Groot, C.J.; van der Valk, P.; Ravid, R.; Biber, K. CX3CL1 and CX3CR1 expression in human brain tissue: Noninflammatory control versus multiple sclerosis. J. Neuropathol. Exp. Neurol. 2003, 62, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.; Reczek, D.; Brondyk, W. Capture-stabilize approach for membrane protein SPR assays. Sci. Rep. 2014, 4, 7360. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment screening by SPR and advanced application to GPCRs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.K.; Fong, A.M.; Swain, P.A.; Chen, S.; Yu, Y.R.; Salafranca, M.N.; Greenleaf, W.B.; Imai, T.; Patel, D.D. Mutational analysis of the fractalkine chemokine domain. Basic amino acid residues differentially contribute to CX3CR1 binding, signaling, and cell adhesion. J. Biol. Chem. 2001, 276, 21632–21641. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Heitman, L.H.; IJzerman, A.P. Kinetic Aspects of the Interaction between Ligand and G Protein-Coupled Receptor: The Case of the Adenosine Receptors. Chem. Rev. 2017, 117, 38–66. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gessesse, B.; Nagaike, T.; Nagata, K.; Shimizu, Y.; Ueda, T. G-Protein Coupled Receptor Protein Synthesis on a Lipid Bilayer Using a Reconstituted Cell-Free Protein Synthesis System. Life 2018, 8, 54. https://doi.org/10.3390/life8040054
Gessesse B, Nagaike T, Nagata K, Shimizu Y, Ueda T. G-Protein Coupled Receptor Protein Synthesis on a Lipid Bilayer Using a Reconstituted Cell-Free Protein Synthesis System. Life. 2018; 8(4):54. https://doi.org/10.3390/life8040054
Chicago/Turabian StyleGessesse, Belay, Takashi Nagaike, Koji Nagata, Yoshihiro Shimizu, and Takuya Ueda. 2018. "G-Protein Coupled Receptor Protein Synthesis on a Lipid Bilayer Using a Reconstituted Cell-Free Protein Synthesis System" Life 8, no. 4: 54. https://doi.org/10.3390/life8040054
APA StyleGessesse, B., Nagaike, T., Nagata, K., Shimizu, Y., & Ueda, T. (2018). G-Protein Coupled Receptor Protein Synthesis on a Lipid Bilayer Using a Reconstituted Cell-Free Protein Synthesis System. Life, 8(4), 54. https://doi.org/10.3390/life8040054