Bioinformatic Workflows for Generating Complete Plastid Genome Sequences—An Example from Cabomba (Cabombaceae) in the Context of the Phylogenomic Analysis of the Water-Lily Clade


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Taxon Sampling
2.2. DNA Isolation, Shearing and Size Selection
2.3. Library Preparation and DNA Sequencing
2.4. Genome Assembly
2.4.1. Generating Ordered Intersection of Paired-End Reads
2.4.2. Quality Filtering of Raw Reads
2.4.3. Assembly of Contigs
2.4.4. Manual Assembly of Contigs
2.4.5. Identification and Confirmation of IR Boundaries
2.4.6. Extraction of Plastid Reads
2.4.7. Assembly Quality Statistics and Genome Maps
2.5. Genome Annotation
2.5.1. Generating Raw Gene Annotations
2.5.2. Manual Correction of Annotations
2.6. Sequence Alignment
2.6.1. Extraction and Alignment of Coding Regions
2.6.2. Removal of Gap Positions
2.7. Phylogenetic Analysis
2.8. Overview of Workflow and Tutorial
3. Results
3.1. Genome Assembly
3.2. Genome Structure and IR Length
3.3. Gene Content
3.4. Alignment Statistics
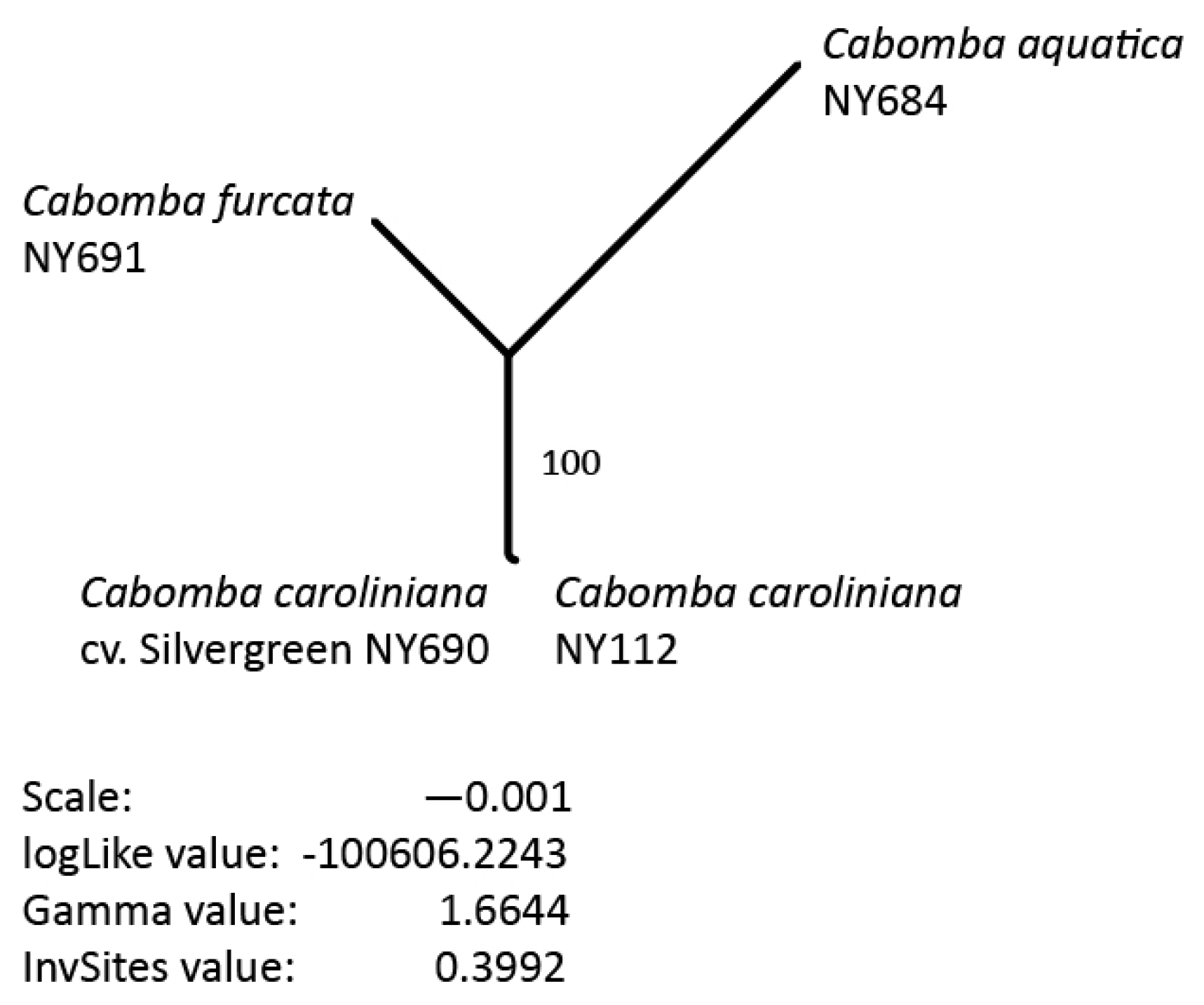
3.5. Phylogenetic Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, L.; Su, Y.J.; Wang, T. Plastid genome sequencing, comparative genomics, and phylogenomics: Current status and prospects. J. Syst. Evol. 2010, 48, 77–93. [Google Scholar] [CrossRef]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Sun, L.; Penny, D. The origin of land plants: A phylogenomic perspective. Evol. Bioinform. 2015, 11, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.G.; Barrett, C.F.; Soto Gomez, M.; Lam, V.K.Y.; Henriquez, C.L.; Les, D.H.; Davis, J.I.; Cuenca, A.; Petersen, G.; Seberg, O.; et al. Plastid phylogenomics and molecular evolution of Alismatales. Cladistics 2016, 32, 160–178. [Google Scholar] [CrossRef]
- Zhang, N.; Wen, J.; Zimmer, E.A. Another look at the phylogenetic position of the grape order Vitales: Chloroplast phylogenomics with an expanded sampling of key lineages. Mol. Phylogenet. Evol. 2016, 101, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Gruenstaeudl, M.; Nauheimer, L.; Borsch, T. Plastid genome structure and phylogenomics of Nymphaeales: Conserved gene order and new insights into relationships. Plant Syst. Evol. 2017, 303, 1251–1270. [Google Scholar] [CrossRef]
- Ma, P.F.; Zhang, Y.X.; Zeng, C.X.; Guo, Z.H.; Li, D.Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hu, Q.; Al-Shehbaz, I.A.; Luo, X.; Zeng, T.; Guo, X.; Liu, J. Species Delimitation and Interspecific Relationships of the Genus Orychophragmus (Brassicaceae) Inferred from Whole Chloroplast Genomes. Front. Plant Sci. 2016, 7, 1826. [Google Scholar] [CrossRef] [PubMed]
- Spooner, D.M.; Ruess, H.; Iorizzo, M.; Senalik, D.; Simon, P. Entire plastid phylogeny of the carrot genus (Daucus, Apiaceae): Concordance with nuclear data and mitochondrial and nuclear DNA insertions to the plastid. Am. J. Bot. 2017, 104, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Njuguna, W.; Liston, A.; Cronn, R.; Ashman, T.L.; Bassil, N. Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Mol. Phylogenet. Evol. 2013, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Imran, Q.M.; Kang, S.M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Perdereau, A.; Klaas, M.; Barth, S.; Hodkinson, T.R. Plastid genome sequencing reveals biogeographic structure and extensive population genetic variation in wild populations of Phalaris arundinacea L. in north western Europe. GCB Bioenergy 2016, 9, 46–56. [Google Scholar] [CrossRef]
- Welch, A.J.; Collins, K.; Ratan, A.; Drautz-Moses, D.I.; Schuster, S.C.; Lindqvist, C. The quest to resolve recent radiations: Plastid phylogenomics of extinct and endangered Hawaiian endemic mints (Lamiaceae). Mol. Phylogenet. Evol. 2016, 99, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Bakker, F.T.; Lei, D.; Yu, J.; Mohammadin, S.; Wei, Z.; van de Kerke, S.; Gravendeel, B.; Nieuwenhuis, M.; Staats, M.; Alquezar-Planas, D.E.; et al. Herbarium genomics: Plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Biol. J. Linn. Soc. 2016, 117, 33–43. [Google Scholar] [CrossRef]
- Mower, J.P.; Vickrey, T.L. Structural Diversity among Plastid Genomes of Land Plants, 1st ed.; Elsevier Ltd.: New York, NY, USA, 2017; ISBN 9780128134573. [Google Scholar]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Staats, M.; Erkens, R.H.J.; van de Vossenberg, B.; Wieringa, J.J.; Kraaijeveld, K.; Stielow, B.; Geml, J.; Richardson, J.E.; Bakker, F.T. Genomic treasure troves: Complete genome sequencing of herbarium and insect museum specimens. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Choi, K.S.; Jansen, R.K. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol. Biol. Evol. 2005, 22, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.W.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Helmy, M.; Awad, M.; Mosa, K.A. Limited resources of genome sequencing in developing countries: Challenges and solutions. Appl. Transl. Genom. 2016, 9, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Myers, R.M. Advancements in next-generation sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, 2011–2013. [Google Scholar] [CrossRef] [PubMed]
- Twyford, A.D.; Ness, R.W. Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Twyford, A.D.; Ennos, R.A. Next-generation hybridization and introgression. Heredity 2012, 108, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Cascales, J.; Bracco, M.; Garberoglio, M.; Poggio, L.; Gottlieb, A. Integral Phylogenomic Approach over Ilex L. Species from Southern South America. Life 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Taylor, J. Next-generation sequencing data interpretation: Enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012, 13, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.; Alessandri, L.; Panero, R.; Arigoni, M.; Olivero, M.; Cordero, F.; Beccuti, M.; Calogero, R.A. Reproducible Bioinformatics Project: A community for reproducible bioinformatics analysis pipelines. bioRxiv 2017, 239947. [Google Scholar] [CrossRef]
- Magoc, T.; Pabinger, S.; Canzar, S.; Liu, X.; Su, Q.; Puiu, D.; Tallon, L.J.; Salzberg, S.L. GAGE-B: An evaluation of genome assemblers for bacterial organisms. Bioinformatics 2013, 29, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.S.; Pyzh, R.; Jeon, M.S.; Amaro, C.; Roig, F.J.; Baker-Austin, C.; Oliver, J.D.; Gibas, C.J. Impact of analytic provenance in genome analysis. BMC Genom. 2014, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, S.; Khan, F.Z.; Lonie, A.; Sinnott, R.O. Investigating reproducibility and tracking provenance—A genomic workflow case study. BMC Bioinform. 2017, 18, 337. [Google Scholar] [CrossRef] [PubMed]
- Orgaard, M. The genus Cabomba (Cabombaceae)—A taxonomic study. Nord. J. Bot. 1991, 11, 179–203. [Google Scholar] [CrossRef]
- De Lima, C.T.; dos Santos, F.d.A.R.; Giulietti, A.M. Morphological strategies of Cabomba (Cabombaceae), a genus of aquatic plants. Acta Bot. Bras. 2014, 28, 327–338. [Google Scholar] [CrossRef]
- McCracken, A.; Bainard, J.D.; Miller, M.C.; Husband, B.C. Pathways of introduction of the invasive aquatic plant Cabomba caroliniana. Ecol. Evol. 2013, 3, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.E.; Darbyshire, S.J.; Jones, R. The Biology of Invasive Alien Plants in Canada. 7. Cabomba caroliniana A. Gray. Can. J. Plant Sci. 2007, 87, 615–638. [Google Scholar] [CrossRef]
- Jacobs, M.J.; Macisaac, H.J. Modelling spread of the invasive macrophyte Cabomba caroliniana. Freshw. Biol. 2009, 54, 296–305. [Google Scholar] [CrossRef]
- Heng, L. bioawk, Version 20110810. Available online: https://github.com/lh3/bioawk (accessed on 28 February 2018).
- Ramey, C.; Fox, B. Bash Reference Manual: Reference Documentation for Bash Edition 4.4; Free Software Foundation: Boston, MA, USA, 2016; ISBN 978-0-9541617-7-4. [Google Scholar]
- Gordon, A. FASTX Toolkit, Version 0.0.14. Available online: https://github.com/agordon/fastx_toolkit (accessed on 28 February 2018).
- Bushnell, B. BBTools Software Package, Version 33.89. Available online: http://sourceforge.net/projects/bbmap (accessed on 28 February 2018).
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Hirsch-Ernst, K.I.; Wolfl, S.; Hellwig, F.H. The chloroplast genome of Nymphaea alba: Whole-genome analyses and the problem of identifying the most basal angiosperm. Mol. Biol. Evol. 2004, 21, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Moore, B.; Batchelor, C.; Salas, F.; Cunningham, F.; Marth, G.T.; Stein, L.; Flicek, P.; Yandell, M.; Eilbeck, K. A standard variation file format for human genome sequences. Genome Biol. 2010, 11, R88. [Google Scholar] [CrossRef] [PubMed]
- Python Software Foundation. Python Language Reference, Version 2.7. Available online: http://www.python.org (accessed on 6 April 2018).
- Perl Development Community. Perl Language Reference, Version 5.26.1. Available online: http://www.perl.org/ (accessed on 6 April 2018).
- Blankenberg, D.; Von Kuster, G.; Bouvier, E.; Baker, D.; Afgan, E.; Stoler, N.; the Galaxy Team; Taylor, J.; Nekrutenko, A. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014, 15, 403. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing, Version 3.4.4. R Foundation for Statistical Computing, Vienna, Austria. Available online: http://www.R-project.org/ (accessed on 30 April 2018).
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Galati, B.G.; Gotelli, M.M.; Rosenfeldt, S.; Lattar, E.C.; Tourn, G.M. Chloroplast dimorphism in leaves of Cabomba caroliniana (Cabombaceae). Aquat. Bot. 2015, 121, 46–51. [Google Scholar] [CrossRef]
- Vialette-Guiraud, A.C.M.; Alaux, M.; Legeai, F.; Finet, C.; Chambrier, P.; Brown, S.C.; Chauvet, A.; Magdalena, C.; Rudall, P.J.; Scutt, C.P. Cabomba as a model for studies of early angiosperm evolution. Ann. Bot. 2011, 108, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Pop, M.; Salzberg, S.L. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008, 24, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Oakley, T.H.; Alexandrou, M.A.; Ngo, R.; Pankey, M.; Churchill, C.K.C.; Chen, W.; Lopker, K.B. Osiris: Accessible and reproducible phylogenetic and phylogenomic analyses within the Galaxy workflow management system. BMC Bioinf. 2014, 15, 230. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv 2018, 256479. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, F.; Rohlsen, D.; Bi, C.; Wang, C.; Xu, Y.; Wei, S.; Ye, Q.; Yin, T.; Ye, N. Organellar genome assembly methods and comparative analysis of horticultural plants. Hortic. Res. 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- McKain, M.R.; Hartsock, R.H.; Wohl, M.M.; Kellogg, E.A. Verdant: Automated annotation, alignment and phylogenetic analysis of whole chloroplast genomes. Bioinformatics 2017, 33, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.R.; Frampton, M.B. Tools and techniques for computational reproducibility. Gigascience 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Leipzig, J. A review of bioinformatic pipeline frameworks. Brief. Bioinform. 2017, 18, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten simple rules for reproducible computational research. PLoS Comput. Biol. 2013, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Preeyanon, L.; Brown, C.T.; Black Pyrkosz, A. Reproducible bioinformatics research for biologists. In Implementing Reproducible Research, 1st ed.; Stodden, V., Leisch, F., Peng, R.D., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 185–217. ISBN 978-1-4665615-9-5. [Google Scholar]



{kind=link}
{kind=link}
| Species Name | Publication of Genome Sequence | GenBank Accession Number | Sample ID | Herbarium Voucher |
|---|---|---|---|---|
| C. aquatica Aubl. | this study | MG720559 | NY684 | Gartenherbarbeleg Cubr 50791 (B) |
| C. caroliniana A. Gray | Gruenstaeudl et al. (2017) | KT705317 | NY112 | J.C. Ludwig s.n. (VPI) |
| C. caroliniana cv. ‘Silvergreen’ | this study | MG720558 | NY690 | Gartenherbarbeleg Cubr 50793 (B) |
| C. furcata Gardner | this study | MG967470 | NY691 | Gartenherbarbeleg Cubr 50792 (B) |
| Analysis Step | Details of Analysis | Custom Software Script | Third-Party Software Tool Employed 1 | Computation Time (Mean) |
|---|---|---|---|---|
| Quality control of reads | Generating ordered intersection of R1/R2 reads | Script 01 | bioawk v.20110810 | 6 min 29 s |
| Filtering of reads by quality score | Script 02 | FASTX Toolkit v.0.0.14 | 14 min 03 s | |
| Genome assembly | Assembling reads to contigs | Script 03 | IOGA v.20160910 Python v.2.7.14 | 17 min 12 s |
| Stitching contigs to complete genomes | Manual step | Geneious v.10.2.3 | ||
| Evaluation of assembly | Confirming the IR boundaries | Script 04 | BLAST+ v.2.4.0 | <1 s |
| Extracting reads that map to final assembly | Script 05 | bowtie2 v.2.3.2 samtools v.1.7 BEDtools v.2.27.1 | 3 min 37 s | |
| Generating assembly statistics | Script 06 | bowtie2 v.2.3.2 samtools v.1.7 BEDtools v.2.27.1 | 7 min 20 s | |
| Genome annotation | Generating raw annotations | Manual step | DOGMA cpGAVAS | |
| Converting annotations of DOGMA to GFF format | Script 07 | n.a. | <1 s | |
| Combining annotations of DOGMA and cpGAVAS | Script 08 | Python v.2.7.14 Perl v.5.26.1 | <1 s | |
| Evaluation of annotations | Confirming the validity of annotations | Manual step | Geneious v.10.2.3 | |
| Sequence alignment | Extraction and alignment of coding regions | Script 09 | Python v.2.7.14 | 39 s |
| Removal of gap positions | Script 10 | statAl v.1.4.rev22 trimAl v.1.4.rev22 | <1 s | |
| Phylogenetic analysis | Phylogenetic tree inference under ML, including bootstrapping | Script 11 | R v.3.4.4 | 17 s |
| C. aquatica | C. caroliniana cv. ‘Silvergreen’ | C. furcata | |
|---|---|---|---|
| N of read pairs after quality filtering | 1,896,979 | 4,005,075 | 4,132,180 |
| N of read pairs that mapped to the reference genomes (P of quality-filtered pairs) 1 | 73,452 (3.87%) | 55,254 (1.37%) | 46,681 (1.12%) |
| Mean read length [bp] 2,3 | 593.58 | 595.09 | 594.30 |
| Mean coverage depth [fold] 3 | 349.36 | 266.01 | 224.87 |
| P of bases with coverage depth greater than 20-fold, 50-fold, and 100-fold | 99.89%—99.70%—98.51% | 99.97%—99.93%—90.40% | 99.87%—99.76%—89.72% |
| N of contigs after automatic assembly | 3 | 4 | 3 |
| Size of largest contig [bp] | 89,168 | 80,536 | 90,298 |
| Total length of contigs [bp] | 134,685 | 135,444 | 135,367 |
| N50 [bp] | 89,168 | 80,536 | 90,298 |
| L50 | 1 | 1 | 1 |
| Name of Organism | C. aquatica | C. caroliniana | C. caroliniana cv. ‘Silvergreen’ | C. furcata |
|---|---|---|---|---|
| Genome size (bp) | 159,487 | 164,057 | 160,177 | 160,271 |
| LSC length (bp) | 89,433 | 82,090 | 89,835 | 90,037 |
| SSC length (bp) | 19,114 | 18,827 | 19,392 | 19,384 |
| IR length (bp) | 25,470 | 31,570 | 25,475 | 25,425 |
| N of genes | 116 | 116 | 116 | 116 |
| N of protein-coding genes (duplicated in IR) | 82 (9) | 82 (19) | 82 (9) | 82 (9) |
| N of tRNA genes (duplicated in IR) | 30 (7) | 30 (7) | 30 (7) | 30 (7) |
| N of rRNA genes (duplicated in IR) | 4 (4) | 4 (4) | 4 (4) | 4 (4) |
| Proportion of coding to non-coding regions | 0.69 | 0.69 | 0.69 | 0.69 |
| Average gene density (genes/kb) | 0.85 | 0.89 | 0.85 | 0.85 |
| GC content (%) | 38.0 | 38.3 | 38.0 | 38.1 |
| Statistic | Before Gap Removal | After Gap Removal |
|---|---|---|
| Total alignment length (bp) | 68,922 | 68,451 |
| Average pairwise sequence identity across Cabomba 1 (Pairwise sequence identity of C. caroliniana) | 0.9852 (0.9986) | 0.9891 (0.9998) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruenstaeudl, M.; Gerschler, N.; Borsch, T. Bioinformatic Workflows for Generating Complete Plastid Genome Sequences—An Example from Cabomba (Cabombaceae) in the Context of the Phylogenomic Analysis of the Water-Lily Clade. Life 2018, 8, 25. https://doi.org/10.3390/life8030025
Gruenstaeudl M, Gerschler N, Borsch T. Bioinformatic Workflows for Generating Complete Plastid Genome Sequences—An Example from Cabomba (Cabombaceae) in the Context of the Phylogenomic Analysis of the Water-Lily Clade. Life. 2018; 8(3):25. https://doi.org/10.3390/life8030025
Chicago/Turabian StyleGruenstaeudl, Michael, Nico Gerschler, and Thomas Borsch. 2018. "Bioinformatic Workflows for Generating Complete Plastid Genome Sequences—An Example from Cabomba (Cabombaceae) in the Context of the Phylogenomic Analysis of the Water-Lily Clade" Life 8, no. 3: 25. https://doi.org/10.3390/life8030025
APA StyleGruenstaeudl, M., Gerschler, N., & Borsch, T. (2018). Bioinformatic Workflows for Generating Complete Plastid Genome Sequences—An Example from Cabomba (Cabombaceae) in the Context of the Phylogenomic Analysis of the Water-Lily Clade. Life, 8(3), 25. https://doi.org/10.3390/life8030025