
Has Inositol Played Any Role in the Origin of Life?


{kind=link}
{kind=link}
Abstract
:1. Introduction
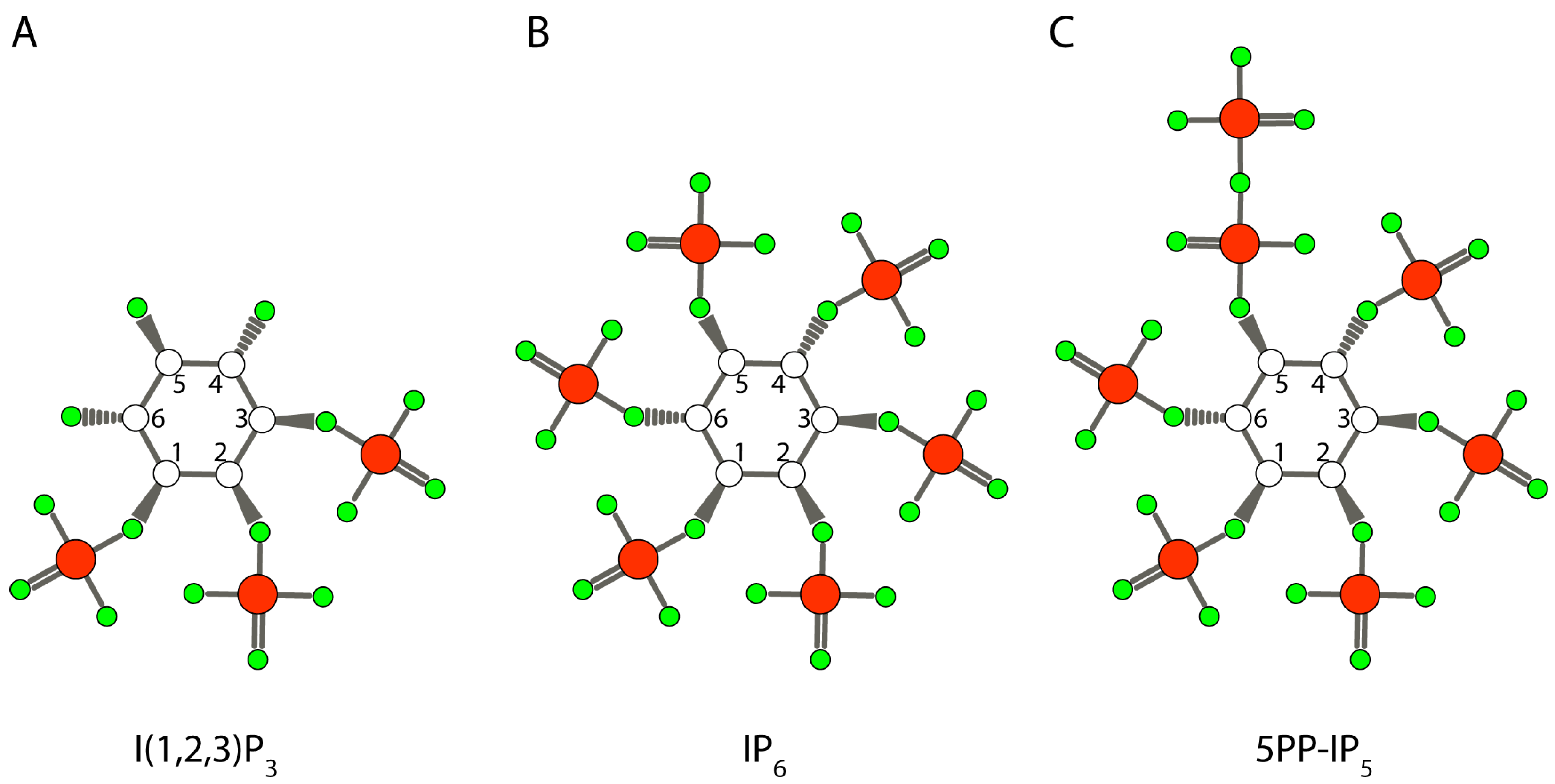
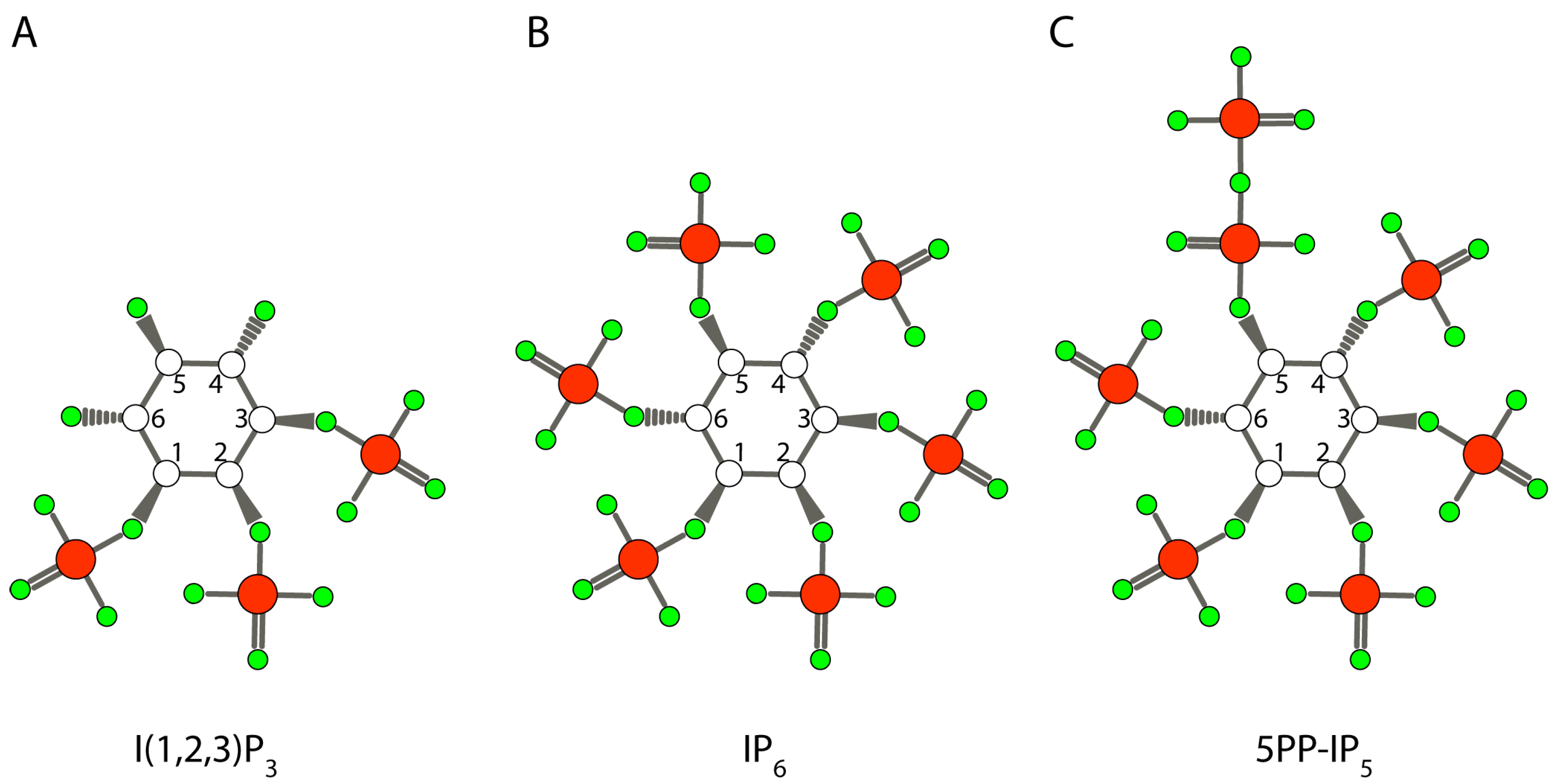
2. Inositol and Inositol Phosphates in Biology Today
3. Inositol Prebiotic Synthesis
4. Inositol and Inositol Phosphates on Early Earth
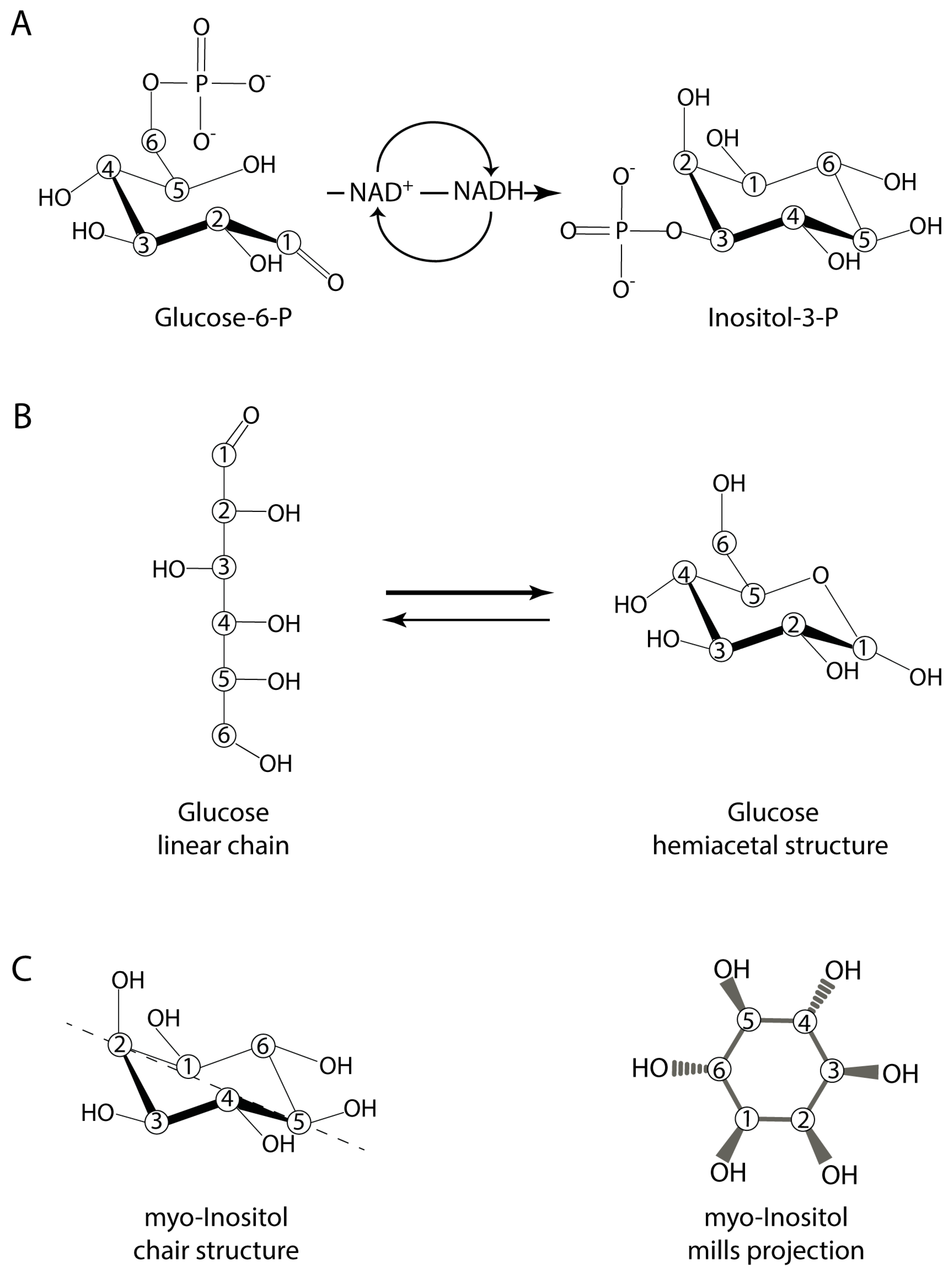
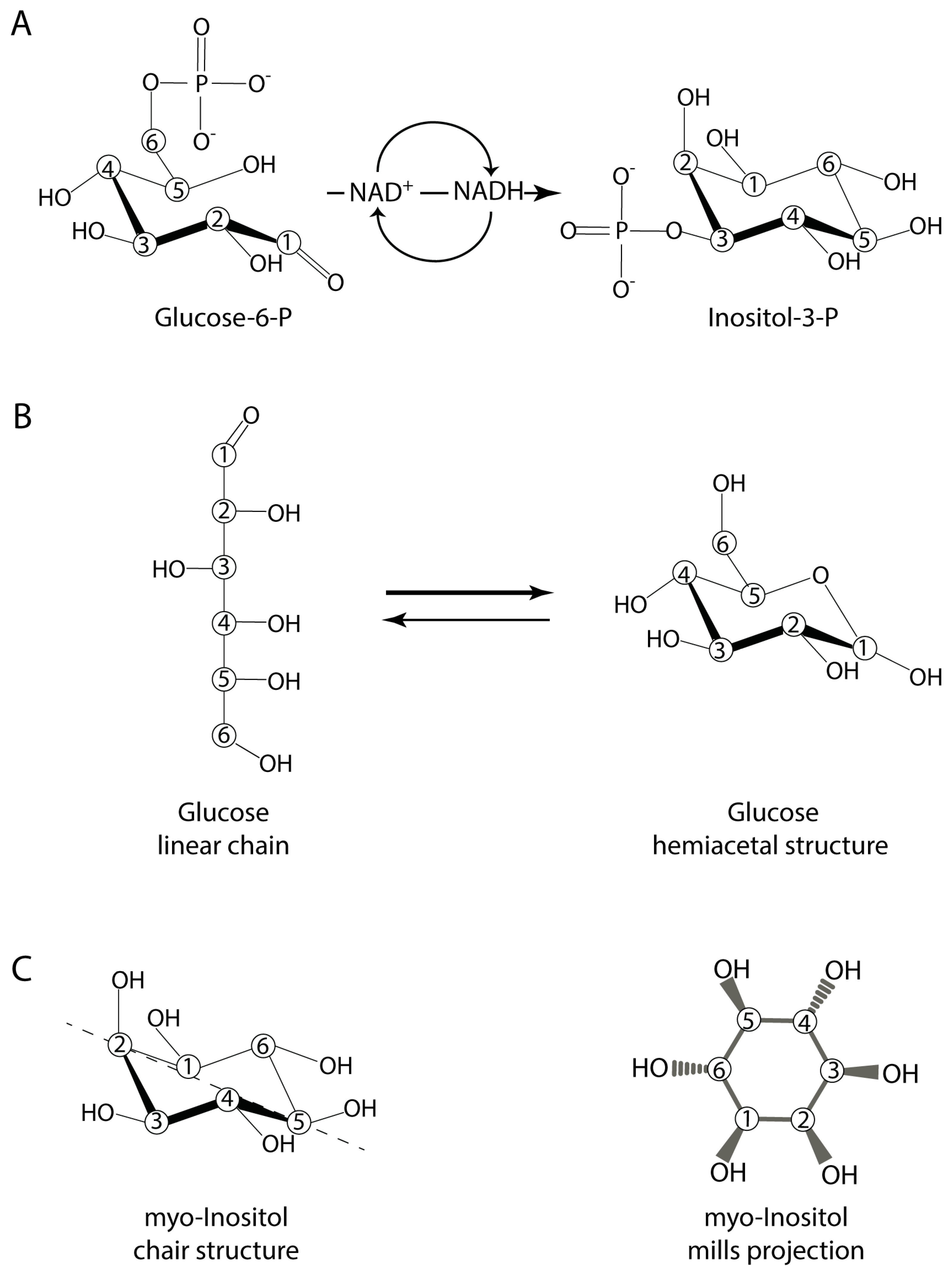
4.1. Inositol and the Initial Organophosphate Molecule
4.2. The Role of the Iron, Magnesium, and Calcium-Chelating Properties of Inositol Phosphate
4.3. Inositol Pyrophosphate as the Initial Phosphorylating Agent
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Westheimer, F.H. Why nature chose phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.; Wellner, A.; Goldin-Azulay, K.; Chabriere, E.; Vorholt, J.A.; Erb, T.J.; Tawfik, D.S. The molecular basis of phosphate discrimination in arsenate-rich environments. Nature 2012, 491, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Wachtershauser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segre, D. Remnants of an ancient metabolism without phosphate. Cell 2017, 168, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Miller, S.L. Are polyphosphates or phosphate esters prebiotic reagents? J. Mol. Evol. 1995, 41, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.; Herschy, B.; Kee, T.P. Phosphorus: A case for mineral-organic reactions in prebiotic chemistry. Orig. Life Evol. Biosph. 2015, 45, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A.; Harnmeijer, J.P.; Buick, R.; Gull, M.; Atlas, Z. Evidence for reactive reduced phosphorus species in the early archean ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 10089–10094. [Google Scholar] [CrossRef] [PubMed]
- Burcar, B.; Pasek, M.; Gull, M.; Cafferty, B.J.; Velasco, F.; Hud, N.V.; Menor-Salvan, C. Darwin’s warm little pond: A one-pot reaction for prebiotic phosphorylation and the mobilization of phosphate from minerals in a urea-based solvent. Angew. Chem. Int. Ed. Engl. 2016, 55, 13249–13253. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.S. Role of magnesium and other divalent cations in ATP-utilizing enzymes. Magnesium 1987, 6, 28–33. [Google Scholar] [PubMed]
- Kazmierczak, J.; Kempe, S.; Kremer, B. Calcium in the Early Evolution of Living Systems: A Biohistorical Approach. Curr. Org. Chem. 2013, 17, 1738–1750. [Google Scholar] [CrossRef]
- Williams, R.J. The evolution of calcium biochemistry. Biochim. Biophys. Acta 2006, 1763, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Saiardi, A. Eukaryotic phosphate homeostasis: The inositol pyrophosphate perspective. Trends Biochem. Sci. 2017, 42, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Livermore, T.M.; Azevedo, C.; Kolozsvari, B.; Wilson, M.S.; Saiardi, A. Phosphate, inositol and polyphosphates. Biochem. Soc. Trans. 2016, 44, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.L.; Chatterjee, A.; Ghosh Dastidar, K.; Majee, M. Diversification and evolution of l-myo-inositol 1-phosphate synthase. FEBS Lett. 2003, 553, 3–10. [Google Scholar] [CrossRef]
- Goncalves, L.G.; Borges, N.; Serra, F.; Fernandes, P.L.; Dopazo, H.; Santos, H. Evolution of the biosynthesis of di-myo-inositol phosphate, a marker of adaptation to hot marine environments. Environ. Microbiol. 2012, 14, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.K.; Novak, J.E.; Agranoff, B.W. Inositol and higher inositol phosphates in neural tissues: Homeostasis, metabolism and functional significance. J. Neurochem. 2002, 82, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.F.; Schell, M.J. Back in the water: The return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Resnick, A.C.; Saiardi, A. Inositol polyphosphates. In Wiley Encyclopedia of Chemical Biology; Wiley: Hoboken, NY, USA, 2008; pp. 1–12. [Google Scholar]
- York, J.D. Regulation of nuclear processes by inositol polyphosphates. Biochim. Biophys. Acta 2006, 1761, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Shears, S.B. Inositol pyrophosphates: Why so many phosphates? Adv. Biol. Regul. 2015, 57, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Wundenberg, T.; Mayr, G.W. Synthesis and biological actions of diphosphoinositol phosphates (inositol pyrophosphates), regulators of cell homeostasis. Biol. Chem. 2012, 393, 979–998. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Livermore, T.M.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhou, C.; Yang, H.; Roberts, M.F. Inositol-1-phosphate synthase from archaeoglobus fulgidus is a class II aldolase. Biochemistry 2000, 39, 12415–12423. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.F. Inositide evolution—Towards turtle domination? J. Physiol. 2005, 566, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Veiga, N.; Torres, J.; Mansell, D.; Freeman, S.; Dominguez, S.; Barker, C.J.; Diaz, A.; Kremer, C. “Chelatable iron pool”: Inositol 1,2,3-trisphosphate fulfils the conditions required to be a safe cellular iron ligand. J. Biol. Inorg. Chem. 2009, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Raboy, V. Myo-inositol-1,2,3,4,5,6-hexakisphosphate. Phytochemistry 2003, 64, 1033–1043. [Google Scholar] [CrossRef]
- Shears, S.B. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal 2001, 13, 151–158. [Google Scholar] [CrossRef]
- Veiga, N.; Torres, J.; Dominguez, S.; Mederos, A.; Irvine, R.F.; Diaz, A.; Kremer, C. The behaviour of myo-inositol hexakisphosphate in the presence of magnesium(II) and calcium(II): Protein-free soluble InsP6 is limited to 49 microm under cytosolic/nuclear conditions. J. Inorg. Biochem. 2006, 100, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Anderson, K.E.; Davidson, K.; Stephens, L.R. Signalling through class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.; Hu, X.; Saiardi, A. Are inositol pyrophosphates signalling molecules? J. Cell Physiol. 2009, 220, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Shears, S.B. Diphosphoinositol polyphosphates: Metabolic messengers? Mol. Pharmacol. 2009, 76, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Szijgyarto, Z.; Bosch, D.; Loss, O.; Azevedo, C.; Saiardi, A. Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J. Biol. Chem. 2011, 286, 31966–31974. [Google Scholar] [CrossRef] [PubMed]
- Wild, R.; Gerasimaite, R.; Jung, J.Y.; Truffault, V.; Pavlovic, I.; Schmidt, A.; Saiardi, A.; Jessen, H.J.; Poirier, Y.; Hothorn, M.; et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 2016, 352, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Onnebo, S.M.; Azevedo, C.; Saiardi, A. Inositol pyrophosphates: Metabolism and signaling. Cell. Mol. Life Sci. 2006, 63, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; DeRose, E.F.; London, R.E.; Shears, S.B. IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nat. Commun. 2014, 5, 4178. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Saiardi, A. Functions of inorganic polyphosphates in eukaryotic cells: A coat of many colours. Biochem. Soc. Trans. 2014, 42, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Rao, N.N.; Ault-Riche, D. Inorganic polyphosphate: A molecule of many functions. Annu. Rev. Biochem. 1999, 68, 89–125. [Google Scholar] [CrossRef] [PubMed]
- Gerasimaite, R.; Pavlovic, I.; Capolicchio, S.; Hofer, A.; Schmidt, A.; Jessen, H.J.; Mayer, A. Inositol pyrophosphate specificity of the SPX-dependent polyphosphate polymerase VTC. ACS Chem. Biol. 2017, 12, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.N.; Docampo, R. Polyphosphate and its diverse functions in host cells and pathogens. PLoS Pathog. 2013, 9, e1003230. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Livermore, T.; Saiardi, A. Protein polyphosphorylation of lysine residues by inorganic polyphosphate. Mol. Cell 2015, 58, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Wholey, W.Y.; Wagner, N.O.; Cremers, C.M.; Mueller-Schickert, A.; Hock, N.T.; Krieger, A.G.; Smith, E.M.; Bender, R.A.; Bardwell, J.C.; et al. Polyphosphate is a primordial chaperone. Mol. Cell 2014, 53, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Kornberg, A. Inorganic polyphosphate in the origin and survival of species. Proc. Natl. Acad. Sci. USA 2004, 101, 16085–16087. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A. Inorganic polyphosphate: Toward making a forgotten polymer unforgettable. J. Bacteriol. 1995, 177, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, Y.; Watanabe, H.; Saitoh, M.; Namba, T. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature 1991, 352, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Saiardi, A.; Ahmadibeni, Y.; Snowman, A.M.; Resnick, A.C.; Kristiansen, T.Z.; Molina, H.; Pandey, A.; Werner, J.K., Jr.; Juluri, K.R.; et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc. Natl. Acad. Sci. USA 2007, 104, 15305–15310. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Bhandari, R.; Resnick, A.C.; Snowman, A.M.; Snyder, S.H. Phosphorylation of proteins by inositol pyrophosphates. Science 2004, 306, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Vera, M.; Lique, F.; Dumouchel, F.; Klos, J.; Rubayo Soneira, J.; Senent, M.L. Cyanide/isocyanide abundances in the interstellar medium-II. Inelastic rate coefficients of Al and Mg compounds. Mon. Not. R. Astron. Soc. 2013, 432, 468–477. [Google Scholar] [CrossRef]
- Costanzo, G.; Saladino, R.; Crestini, C.; Ciciriello, F.; Di Mauro, E. Formamide as the main building block in the origin of nucleic acids. BMC Evol. Biol. 2007, 7 (Suppl. S2), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Costanzo, G.; Negri, R.; Di Mauro, E. A possible prebiotic synthesis of purine, adenine, cytosine, and 4(3H)-pyrimidinone from formamide: Implications for the origin of life. Bioorg. Med. Chem. 2001, 9, 1249–1253. [Google Scholar] [CrossRef]
- Schoffstall, A.M. Prebiotic phosphorylation of nucleosides in formamide. Orig. Life 1976, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Carota, E.; Botta, G.; Kapralov, M.; Timoshenko, G.N.; Rozanov, A.Y.; Krasavin, E.; Di Mauro, E. Meteorite-catalyzed syntheses of nucleosides and of other prebiotic compounds from formamide under proton irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E2746–E2755. [Google Scholar] [CrossRef] [PubMed]
- Shigemasa, Y. Formose reactions: IX. Selective formation of branched sugar alcohols in a modified formose reaction and factors affecting the selectivity. J. Catal. 1980, 62, 107–116. [Google Scholar] [CrossRef]
- Appayee, C.; Breslow, R. Deuterium studies reveal a new mechanism for the formose reaction involving hydride shifts. J. Am. Chem. Soc. 2014, 136, 3720–3723. [Google Scholar] [CrossRef] [PubMed]
- Agranoff, B.W. Turtles all the way: Reflections on myo-inositol. J. Biol. Chem. 2009, 284, 21121–21126. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.; Kimmich, N.; Belisle, W.; Sarinana, J.; Brabham, K.; Garrel, L. Carbonaceous meteorites as a source of sugar-related organic compounds for the early earth. Nature 2001, 414, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Weber, A.L. Bioenergetics and life’s origins. Cold Spring Harb. Perspect. Biol. 2010, 2, a004929. [Google Scholar] [CrossRef] [PubMed]
- Gull, M.; Mojica, M.A.; Fernandez, F.M.; Gaul, D.A.; Orlando, T.M.; Liotta, C.L.; Pasek, M.A. Nucleoside phosphorylation by the mineral schreibersite. Sci. Rep. 2015, 5, 17198. [Google Scholar] [CrossRef] [PubMed]
- Kee, T.P.; Bryant, D.E.; Herschy, B.; Marriott, K.E.; Cosgrove, N.E.; Pasek, M.A.; Atlas, Z.D.; Cousins, C.R. Phosphate activation via reduced oxidation state phosphorus (P). Mild routes to condensed-P energy currency molecules. Life 2013, 3, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Ponnamperuma, C.; Mack, R. Nucleotide synthesis under possible primitive earth conditions. Science 1965, 148, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Gull, M.; Zhou, M.; Fernandez, F.M.; Pasek, M.A. Prebiotic phosphate ester syntheses in a deep eutectic solvent. J. Mol. Evol. 2014, 78, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Resnick, A.C.; Saiardi, A. Inositol polyphosphate multikinase: Metabolic architect of nuclear inositides. Front. Biosci. 2008, 13, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Cockcroft, S. Human ITPK1: A reversible inositol phosphate kinase/phosphatase that links receptor-dependent phospholipase C to Ca2+-activated chloride channels. Sci. Signal 2008, 1, pe5. [Google Scholar] [CrossRef] [PubMed]
- Shears, S.B. How versatile are inositol phosphate kinases? Biochem. J. 2004, 377, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Letcher, A.J.; Schell, M.J.; Irvine, R.F. Do mammals make all their own inositol hexakisphosphate? Biochem. J. 2008, 416, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Pisani, F.; Livermore, T.; Rose, G.; Chubb, J.R.; Gaspari, M.; Saiardi, A. Analysis of dictyostelium discoideum inositol pyrophosphate metabolism by gel electrophoresis. PLoS ONE 2014, 9, e85533. [Google Scholar] [CrossRef] [PubMed]
- Pizer, L.I.; Ballou, C.E. Specificity of phosphoglyceric acid mutase. J. Biol. Chem. 1959, 234, 1138–1142. [Google Scholar] [PubMed]
- Hawkins, P.T.; Poyner, D.R.; Jackson, T.R.; Letcher, A.J.; Lander, D.A.; Irvine, R.F. Inhibition of iron-catalysed hydroxyl radical formation by inositol polyphosphates: A possible physiological function for myo-inositol hexakisphosphate. Biochem. J. 1993, 294 Pt 3, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.J.; French, P.J.; Moore, A.J.; Nilsson, T.; Berggren, P.O.; Bunce, C.M.; Kirk, C.J.; Michell, R.H. Inositol 1,2,3-trisphosphate and inositol 1,2- and/or 2,3-bisphosphate are normal constituents of mammalian cells. Biochem. J. 1995, 306 Pt 2, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.R.; Hawkins, P.T.; Morris, A.J.; Downes, P.C. l-myo-inositol 1,4,5,6-tetrakisphosphate (3-hydroxy)kinase. Biochem. J. 1988, 249, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.R.; Hawkins, P.T.; Downes, C.P. An analysis of myo-[3H]inositol trisphosphates found in myo-[3H]inositol prelabelled avian erythrocytes. Biochem. J. 1989, 262, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.S.; Rossier, M.F.; Hughes, A.R.; Shears, S.B.; Armstrong, D.L.; Putney, J.W., Jr. Activation of Ca2+ entry into acinar cells by a non-phosphorylatable inositol trisphosphate. Nature 1991, 352, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Dominguez, S.; Cerda, M.F.; Obal, G.; Mederos, A.; Irvine, R.F.; Diaz, A.; Kremer, C. Solution behaviour of myo-inositol hexakisphosphate in the presence of multivalent cations. Prediction of a neutral pentamagnesium species under cytosolic/nuclear conditions. J. Inorg. Biochem. 2005, 99, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Veiga, N.; Torres, J.; Godage, H.Y.; Riley, A.M.; Dominguez, S.; Potter, B.V.; Diaz, A.; Kremer, C. The behaviour of inositol 1,3,4,5,6-pentakisphosphate in the presence of the major biological metal cations. J. Biol. Inorg. Chem. 2009, 14, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Radenberg, T.; Thiel, U.; Vogel, G.; Khoo, K.H.; Dell, A.; Jackson, T.R.; Hawkins, P.T.; Mayr, G.W. The detection, purification, structural characterization, and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). J. Biol. Chem. 1993, 268, 4009–4015. [Google Scholar] [PubMed]
- Hand, C.E.; Honek, J.F. Phosphate transfer from inositol pyrophosphates insP5PP and insP4(PP)2: A semi-empirical investigation. Bioorg. Med. Chem. Lett. 2007, 17, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Fridy, P.C.; Otto, J.C.; Dollins, D.E.; York, J.D. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 2007, 282, 30754–30762. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Erdjument-Bromage, H.; Snowman, A.M.; Tempst, P.; Snyder, S.H. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 1999, 9, 1323–1326. [Google Scholar] [CrossRef]
- Mayr, G.W.; Radenberg, T.; Thiel, U.; Vogel, G.; Stephens, L.R. Phosphoinositol diphosphates: Non-enzymic formation in vitro and occurrence in vivo in the cellular slime mold dictyostelium. Carbohydr. Res. 1992, 234, 247–262. [Google Scholar] [CrossRef]
- Lin, H.; Lindner, K.; Mayr, G.W. Synthesis and nonradioactive micro-analysis of diphosphoinositol phosphates by hplc with postcolumn complexometry. Methods Mol. Biol. 2010, 645, 103–122. [Google Scholar] [PubMed]
- Robertson, M.P.; Joyce, G.F. The origins of the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, a003608. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.M.; Blaber, M. Protein design at the interface of the pre-biotic and biotic worlds. Arch. Biochem. Biophys. 2012, 526, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible archean ocean. Mol. Syst. Biol. 2014, 10, 725. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M. The emergence of patterning in lifes origin and evolution. Int. J. Dev. Biol. 2009, 53, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.R. The minimotif synthesis hypothesis for the origin of life. J. Transl. Sci. 2016, 2, 289–296. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saiardi, A. Has Inositol Played Any Role in the Origin of Life? Life 2017, 7, 24. https://doi.org/10.3390/life7020024
Saiardi A. Has Inositol Played Any Role in the Origin of Life? Life. 2017; 7(2):24. https://doi.org/10.3390/life7020024
Chicago/Turabian StyleSaiardi, Adolfo. 2017. "Has Inositol Played Any Role in the Origin of Life?" Life 7, no. 2: 24. https://doi.org/10.3390/life7020024
APA StyleSaiardi, A. (2017). Has Inositol Played Any Role in the Origin of Life? Life, 7(2), 24. https://doi.org/10.3390/life7020024