
Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
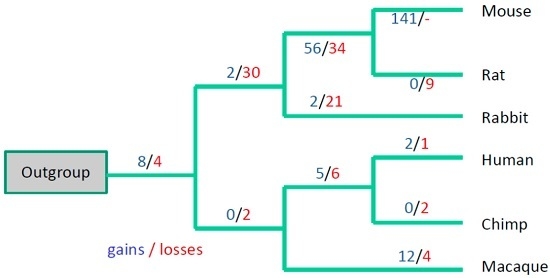
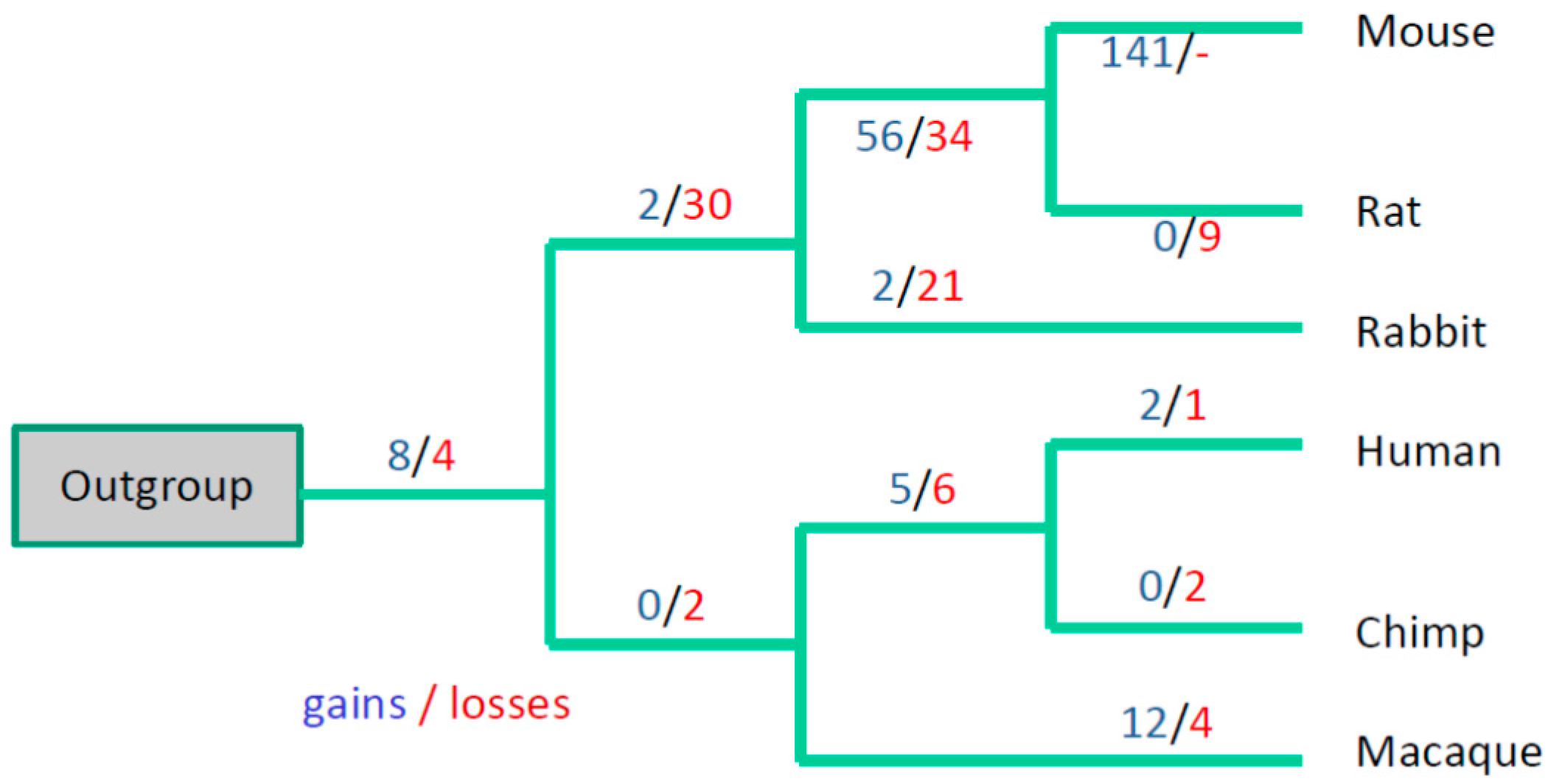
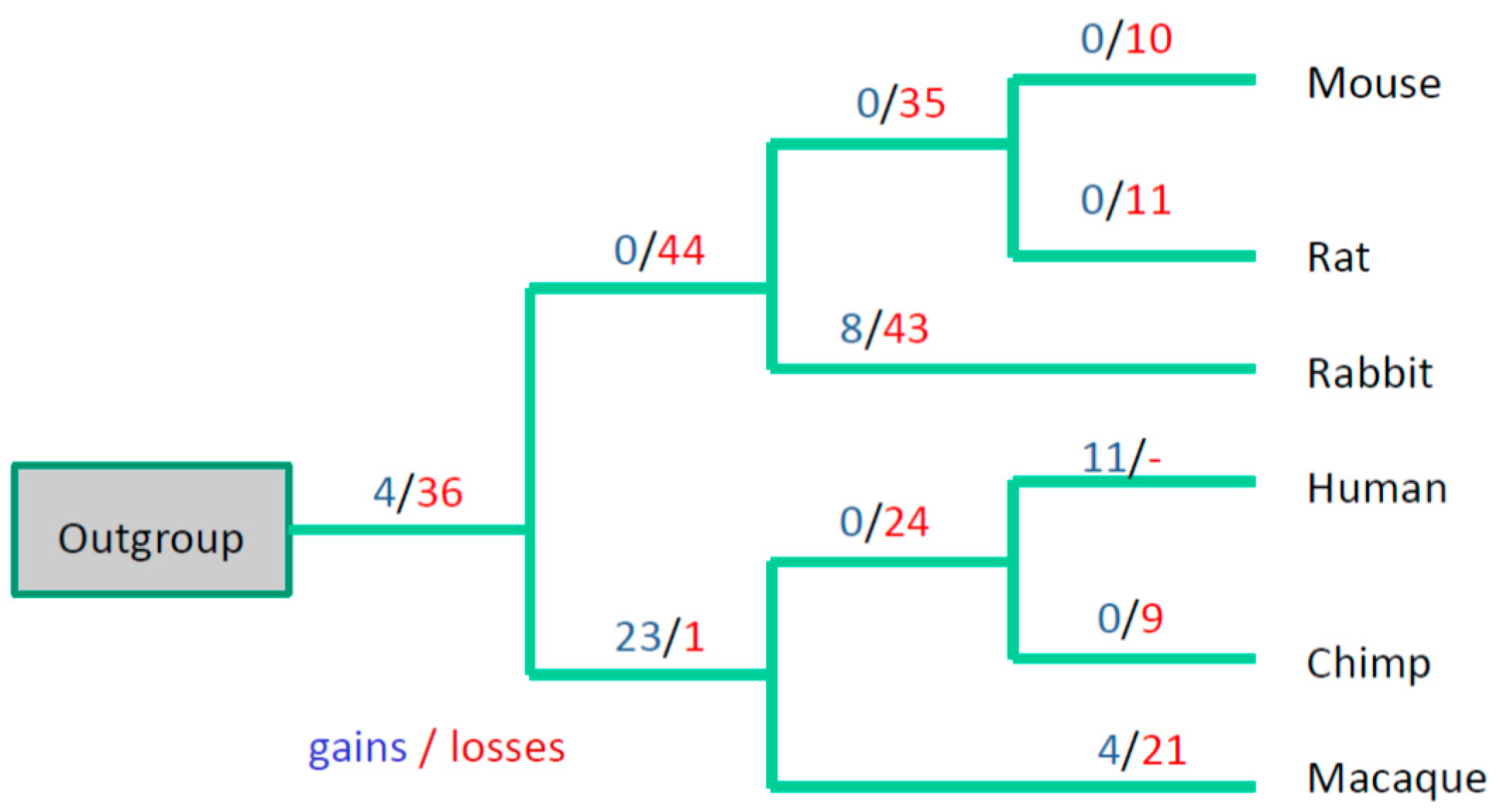
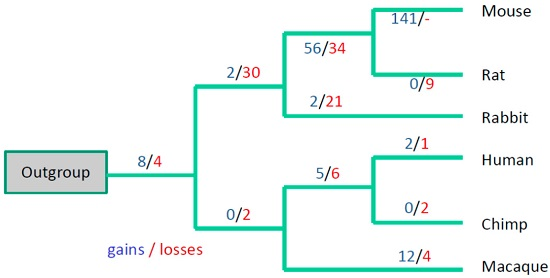
3. Results
3.1. Datasets
3.2. Evolutionary Conservation of Splicing Signals
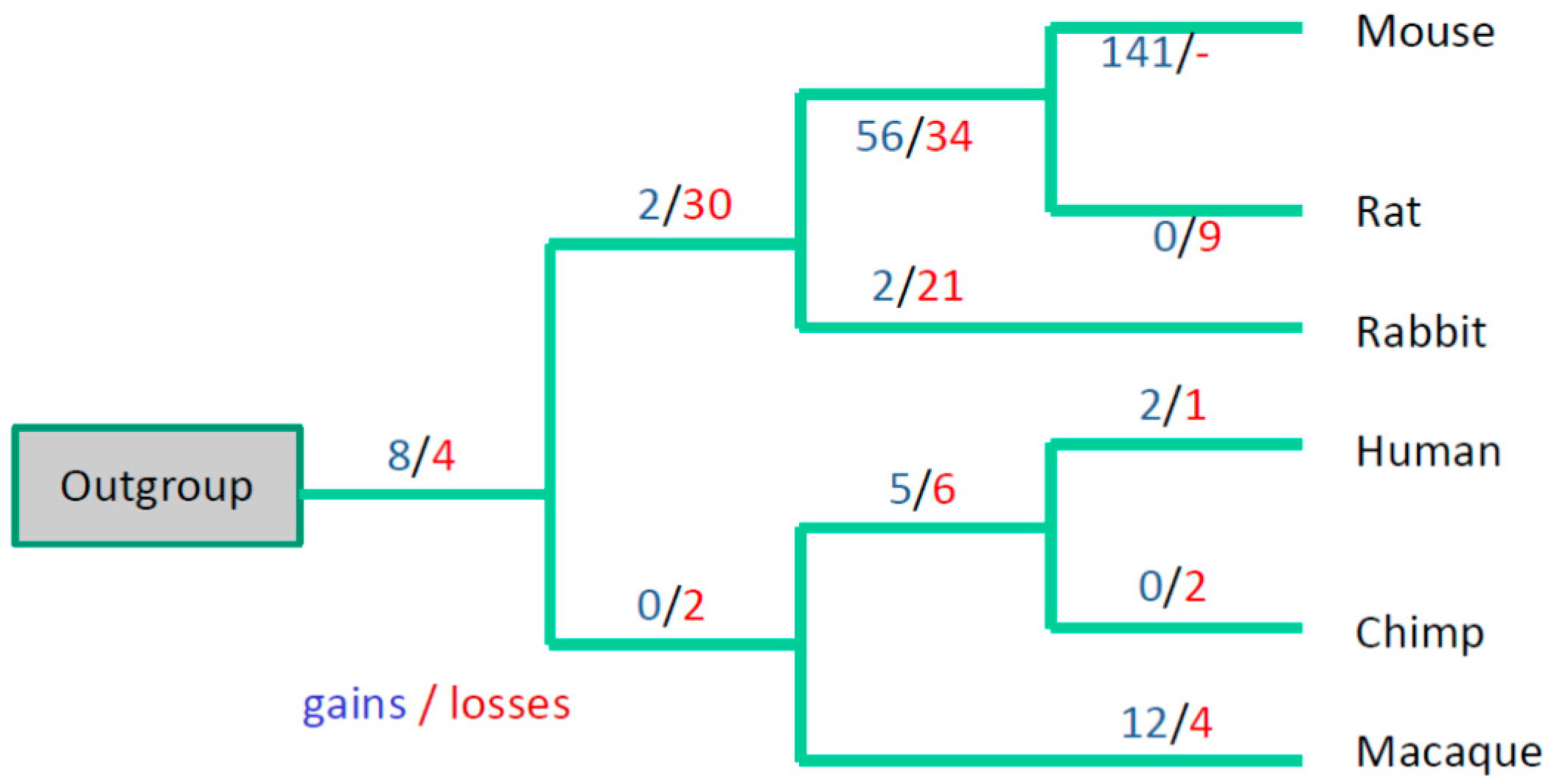
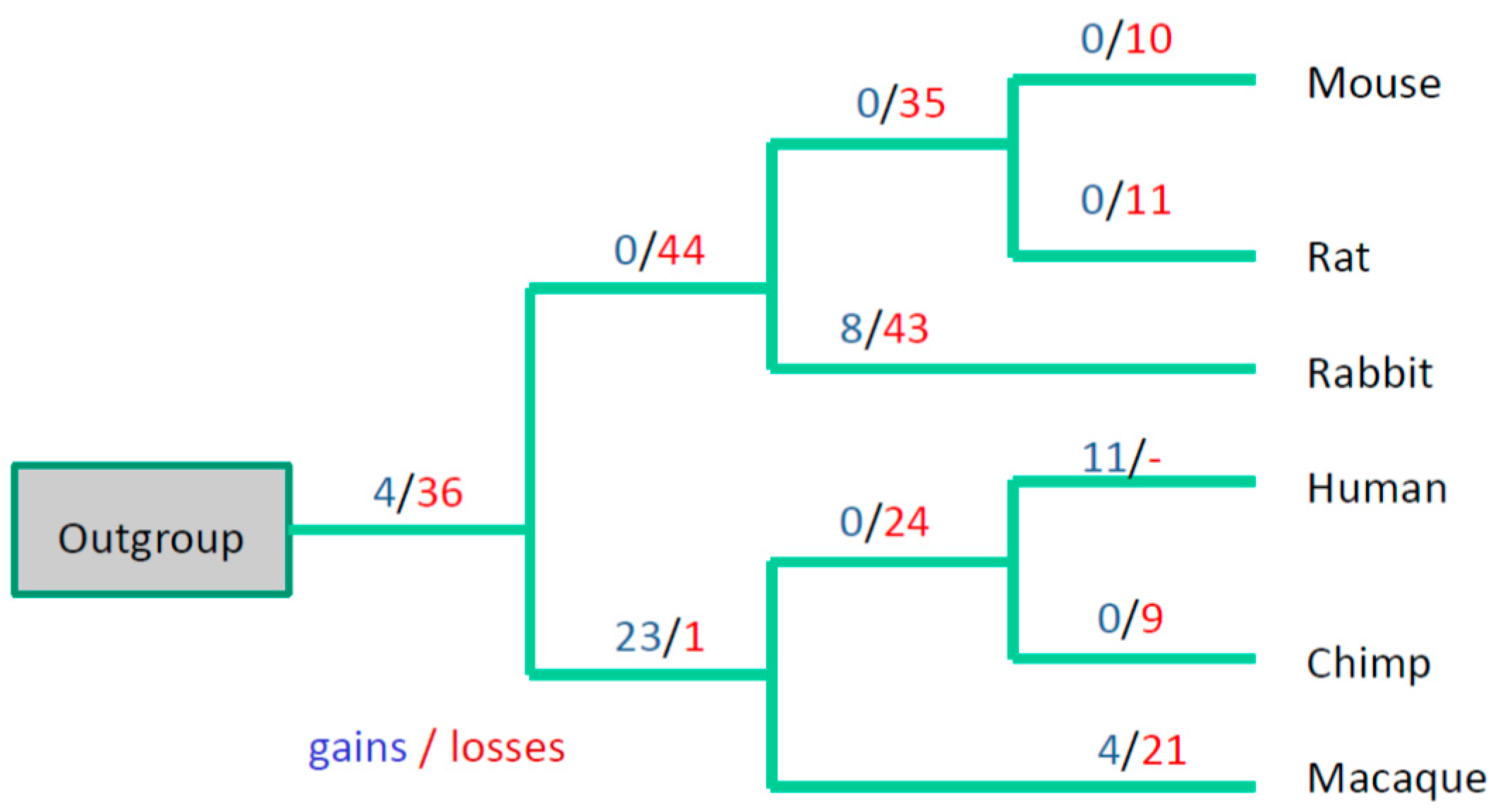
3.3. Evolutionary Conservation of the Exon-Intron Structure
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| lncRNA | long non-coding RNA |
| lincRNA | long intergenic non-coding RNA |
| nt | nucleotide |
References
- Ponting, C.P.; Belgard, T.G. Transcribed dark matter: Meaning or myth? Hum. Mol. Genet. 2010, 19, R162–R168. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.A.; Kugel, J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 2006, 7, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lin, L.; Soh, B.S.; Stanton, L.W. Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 2013, 29, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Van Bakel, H.; Hughes, T.R. Establishing legitimacy and function in the new transcriptome. Brief. Funct. Genom. Proteom. 2009, 8, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. Dark matter transcripts: Sound and fury, signifying nothing? PLoS Biol. 2010, 8, e1000370. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Ponting, C.P. Catalogues of mammalian long noncoding RNAs: Modest conservation and incompleteness. Genome Biol. 2009, 10, R124. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Managadze, D.; Rogozin, I.B.; Chernikova, D.; Shabalina, S.A.; Koonin, E.V. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol. Evol. 2011, 3, 1390–1404. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Chernikova, D.; Rogozin, I.B.; Poliakov, E.; Managadze, D.; Koonin, E.V.; Milanesi, L. Transposable element insertions in long intergenic non-coding RNA genes. Front. Bioeng. Biotechnol. 2015, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Dinger, M.E.; Mattick, J.S. Non-coding RNAs in homeostasis, disease and stress responses: An evolutionary perspective. Brief. Funct. Genom. 2013, 12, 254–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006, 2, e29. [Google Scholar] [CrossRef] [PubMed]
- Managadze, D.; Lobkovsky, A.E.; Wolf, Y.I.; Shabalina, S.A.; Rogozin, I.B.; Koonin, E.V. The vast, conserved mammalian lincRNome. PLoS Comput. Biol. 2013, 9, e1002917. [Google Scholar] [CrossRef] [PubMed]
- Vance, K.W.; Ponting, C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Chodroff, R.A.; Goodstadt, L.; Sirey, T.M.; Oliver, P.L.; Davies, K.E.; Green, E.D.; Molnar, Z.; Ponting, C.P. Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol. 2010, 11, R72. [Google Scholar] [CrossRef] [PubMed]
- Schuler, A.; Ghanbarian, A.T.; Hurst, L.D. Purifying selection on splice-related motifs, not expression level nor RNA folding, explains nearly all constraint on human lincRNAs. Mol. Biol. Evol. 2014, 31, 3164–3183. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Chang, S.C.; Tucker, T.; Thorogood, N.P.; Brown, C.J. Mechanisms of X-chromosome inactivation. Front. Biosci. 2005, 11, 852–866. [Google Scholar] [CrossRef]
- Duret, L.; Chureau, C.; Samain, S.; Weissenbach, J.; Avner, P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 2006, 312, 1653–1655. [Google Scholar] [PubMed]
- Elisaphenko, E.A.; Kolesnikov, N.N.; Shevchenko, A.I.; Rogozin, I.B.; Nesterova, T.B.; Brockdorff, N.; Zakian, S.M. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS ONE 2008, 3, e2521. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Crowe, M.L.; Grimmond, S.M.; Mattick, J.S. NRED: A database of long noncoding RNA expression. Nucleic Acids Res. 2009, 37, D122–D126. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Goecks, J.; Nekrutenko, A.; Taylor, J. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Long, M. Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999, 27, 3219–3228. [Google Scholar] [PubMed]
- Banfai, B.; Jia, H.; Khatun, J.; Wood, E.; Risk, B.; Gundling, W.E., Jr.; Kundaje, A.; Gunawardena, H.P.; Yu, Y.; Xie, L.; et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012, 22, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Russell, P.; Ingolia, N.T.; Weissman, J.S.; Lander, E.S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 2013, 154, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Kutter, C.; Watt, S.; Stefflova, K.; Wilson, M.D.; Goncalves, A.; Ponting, C.P.; Odom, D.T.; Marques, A.C. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.W.; Fedorov, A.; Gilbert, W. Large-scale comparison of intron positions in mammalian genes shows intron loss but no gain. Proc. Natl. Acad. Sci. USA 2003, 100, 7158–7162. [Google Scholar] [CrossRef] [PubMed]
- Csuros, M.; Rogozin, I.B.; Koonin, E.V. A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes. PLoS Comput. Biol. 2011, 7, e1002150. [Google Scholar] [CrossRef] [PubMed]
- Calviello, L.; Mukherjee, N.; Wyler, E.; Zauber, H.; Hirsekorn, A.; Selbach, M.; Landthaler, M.; Obermayer, B.; Ohler, U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 2016, 13, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Carvunis, A.R.; Rolland, T.; Wapinski, I.; Calderwood, M.A.; Yildirim, M.A.; Simonis, N.; Charloteaux, B.; Hidalgo, C.A.; Barbette, J.; Santhanam, B.; et al. Proto-genes and de novo gene birth. Nature 2012, 487, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Rothnagel, J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features of lincRNA Genes | Mouse | Human |
|---|---|---|
| Number of all lincRNAs | 2,390 | 589 |
| Number of intron-containing lincRNAs | 979 | 245 |
| Number of exons | 3,439 | 1,194 |
| Number of introns | 2,462 | 949 |
| Number of exons shorter than 15 nt | 41 | 7 |
| Number of introns per lincRNA | 2.52 | 3.86 |
| Average gene length, nt (standard error) | 11,775 (712) | 17,192 (1,921) |
| Median gene length, nt | 2,535 | 2,626 |
| Average exon length, nt (standard error) | 524 (21) | 409 (48) |
| Median exon length, nt | 464 | 356 |
| Average intron length, nt (standard error) | 9,621 (1,631) | 10,562 (4,539) |
| Median intron length, nt | 2,615 | 2,116 |
| Species | Common Name (Number of Orthologs) | Splice Site Pairwise Comparison with Mouse or Human as a Reference | |||||
|---|---|---|---|---|---|---|---|
| Donor Splicing Site (GT or GC dinucleotide) | Acceptor Splicing Site (AG dinucleotide) | ||||||
| Number of Matches | Number of Mismatches | Percent Matches | Number of Matches | Number of Mismatches | Percent Matches | ||
| Mouse as a reference | |||||||
| Rattus norvegicus | Rat (2285) | 1555 | 569 | 73% | 1448 | 669 | 68% |
| Oryctolagus cuniculus | Rabbit (1522) | 518 | 258 | 67% | 419 | 306 | 58% |
| Homo sapiens | Human (2091) | 902 | 619 | 59% | 746 | 715 | 51% |
| Pan troglodytes | Chimp (2068) | 826 | 606 | 58% | 703 | 692 | 50% |
| Macaca mulatta | Macaque (1971) | 807 | 543 | 60% | 682 | 647 | 51% |
| Bos taurus | Cow (1815) | 694 | 402 | 63% | 560 | 498 | 53% |
| Canis lupus familiaris | Dog (1897) | 714 | 512 | 58% | 627 | 581 | 52% |
| Loxodonta africana | Elephant (1485) | 499 | 247 | 67% | 428 | 312 | 58% |
| Echinops telfairi | Tenrec (1256) | 368 | 179 | 67% | 283 | 193 | 59% |
| Takifugu Rubripes | Fugu (203) | 36 | 28 | 56% | 24 | 28 | 46% |
| Monodelphis domestica | Opossum (1068) | 249 | 169 | 60% | 162 | 150 | 52% |
| Dasypus novemcinctus | Armadillo (1426) | 469 | 260 | 64% | 382 | 322 | 54% |
| Gallus gallus | Chicken (472) | 113 | 36 | 76% | 75 | 43 | 64% |
| Danio rerio | Zebrafish (207) | 44 | 27 | 62% | 26 | 32 | 45% |
| Tetraodon nigroviridis | Tetraodon (226) | 46 | 24 | 66% | 29 | 28 | 51% |
| Xenopus tropicalis | Frog (312) | 74 | 37 | 67% | 51 | 40 | 56% |
| Human as a reference | |||||||
| Pan troglodytes | Chimp (575) | 870 | 19 | 98% | 867 | 15 | 98% |
| Macaca mulatta | Macaque (564) | 800 | 53 | 94% | 828 | 42 | 95% |
| Mus musculus | Mouse (488) | 368 | 120 | 75% | 364 | 105 | 78% |
| Rattus norvegicus | Rat (476) | 369 | 112 | 77% | 342 | 102 | 77% |
| Oryctolagus cuniculus | Rabbit (463) | 445 | 86 | 84% | 415 | 114 | 78% |
| Bos taurus | Cow (527) | 531 | 122 | 81% | 484 | 144 | 77% |
| Canis lupus familiaris | Dog (476) | 546 | 121 | 82% | 543 | 118 | 82% |
| Loxodonta africana | Elephant (458) | 364 | 82 | 82% | 341 | 83 | 80% |
| Echinops telfairi | Tenrec (419) | 196 | 59 | 77% | 175 | 68 | 72% |
| Dasypus novemcinctus | Armadillo (468) | 362 | 95 | 79% | 320 | 122 | 72% |
| Monodelphis domestica | Opossum (287) | 213 | 35 | 86% | 189 | 62 | 75% |
| Gallus gallus | Chicken (131) | 33 | 10 | 77% | 23 | 18 | 56% |
| Takifugu Rubripes | Fugu (80) | 48 | 7 | 87% | 51 | 11 | 82% |
| Danio rerio | Zebrafish (79) | 43 | 7 | 86% | 44 | 9 | 83% |
| Tetraodon nigroviridis | Tetraodon (87) | 49 | 16 | 75% | 52 | 18 | 74% |
| Xenopus tropicalis | Frog (89) | 29 | 4 | 88% | 29 | 10 | 74% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernikova, D.; Managadze, D.; Glazko, G.V.; Makalowski, W.; Rogozin, I.B. Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals. Life 2016, 6, 27. https://doi.org/10.3390/life6030027
Chernikova D, Managadze D, Glazko GV, Makalowski W, Rogozin IB. Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals. Life. 2016; 6(3):27. https://doi.org/10.3390/life6030027
Chicago/Turabian StyleChernikova, Diana, David Managadze, Galina V. Glazko, Wojciech Makalowski, and Igor B. Rogozin. 2016. "Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals" Life 6, no. 3: 27. https://doi.org/10.3390/life6030027
APA StyleChernikova, D., Managadze, D., Glazko, G. V., Makalowski, W., & Rogozin, I. B. (2016). Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals. Life, 6(3), 27. https://doi.org/10.3390/life6030027