Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis

Abstract
:1. Introduction
2. Material and Method
- (1)
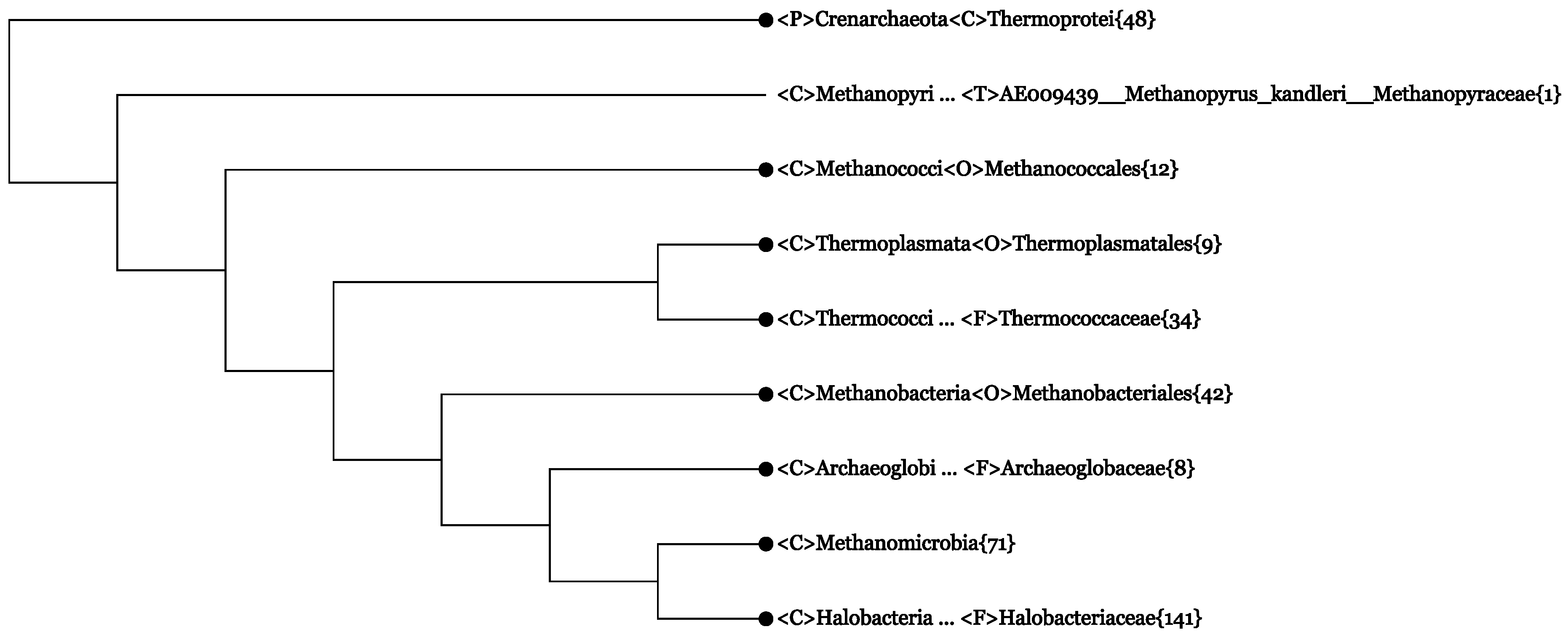
- CVTree3 is equipped with an interactive tree display, which allows collapsing or expanding the tree branches at the disposal of the user. The user may concentrate on an interested taxon by submitting an enquiry; only the neighborhood of the taxon is expanded and all of the rest collapsed properly, keeping the topology unchanged. Here, “collapsing” means replacing a whole branch by a single leaf. Usually, a collapsed branch is labeled by the name of the highest common taxon followed by the number of strains it represents. For example, <C>Methanococci{12} denotes a class-level monophyletic branch containing 12 leaves. If a taxon name is seen in two (or more) collapsed branches, such as <C>Classname{3/12} and <C>Classname{9/12}, then the taxonomically monophyletic class does not correspond to a single branch in the collapsed tree.
- (2)
- The web server reports “convergence statistics” of all tree branches, i.e., a list of all monophyletic and non-monophyletic taxa at all taxonomic ranks for every K-value. For example, the first two lines of the report read:<D>Archaea{165}− − K5K6K7−(Numerals in curly brackets tell the number of organisms present in a collapsed branch.) Therefore, the two domains Archaea and Bacteria are both well defined as monophyletic branches at K = 5 and 6. We note that in the statistics, only genomes with complete lineage information are counted. The example project referred to in this paper contained, in addition, 14 archaeal and 143 bacterial genomes with one or more “unclassified” rank in the lineage. Therefore, in total {165 + 14}= 179 Archaea and {2707+243}= 2850 Bacteria genomes were used. The {m+n}convention is useful for looking for incomplete lineages in CVTree branches.<D>Bacteria{2707} − − K5K6 − −
- (3)
- The lineage information of an organism is given in one line with labels <D>, <P>, <C>, <O>, <F>, <G> and <S>, standing for the ranks domain, phylum, class, order, family, genus and species. The sTrain label <T> does not appear in lineage information, but may be seen in a leaf. The original lineage information of the built-in genomes was taken from the NCBI taxonomy. The lineage information of user’s genomes was provided at uploading. Users are allowed to make lineage modifications and to see new statistics after doing re-collapsing.
- (4)
- When displaying a tree, the user may pull down a lineage modification window and enter a trial lineage in the form “old_lineage new_lineage”. For example, the initial lineage for <T>Caldiarchaeum_cryptofilum_OPF8_uid58601 put it in phylum Thaumarchaeota, but there is evidence that it belongs to a new phylum, Aigarchaeota, so the modification may look like:The modification line is not required to contain all ranks, but the written part must be uniquely recognizable. By submitting the lineage modification, the user performs “re-collapsing” and gets a new report of “convergence statistics”.<P>Thaumarchaeota · · · <G>Caldiarchaeum <P>Aigarchaeota · · · <G>Caldiarchaeum
- (5)
- The user may select any part of a CVTree and produce a print-quality figure in SVG, EPS, PDF or PNG format.
3. Outline of Archaea Taxonomy at and above the Rank Order
4. Results and Discussion
4.1. 16S rRNA Archaeal Phylogeny According to All-Species Living Tree
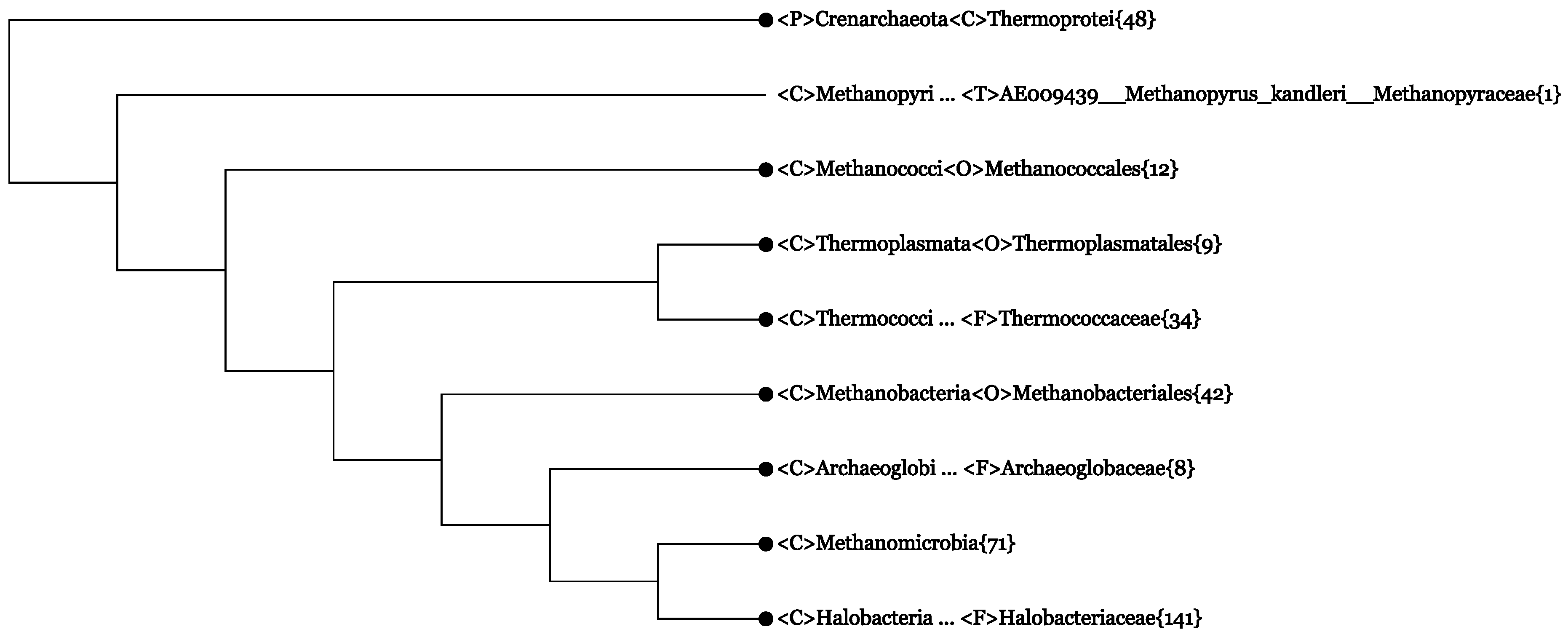
4.2. The Whole-Genome-Based CVTree Phylogeny
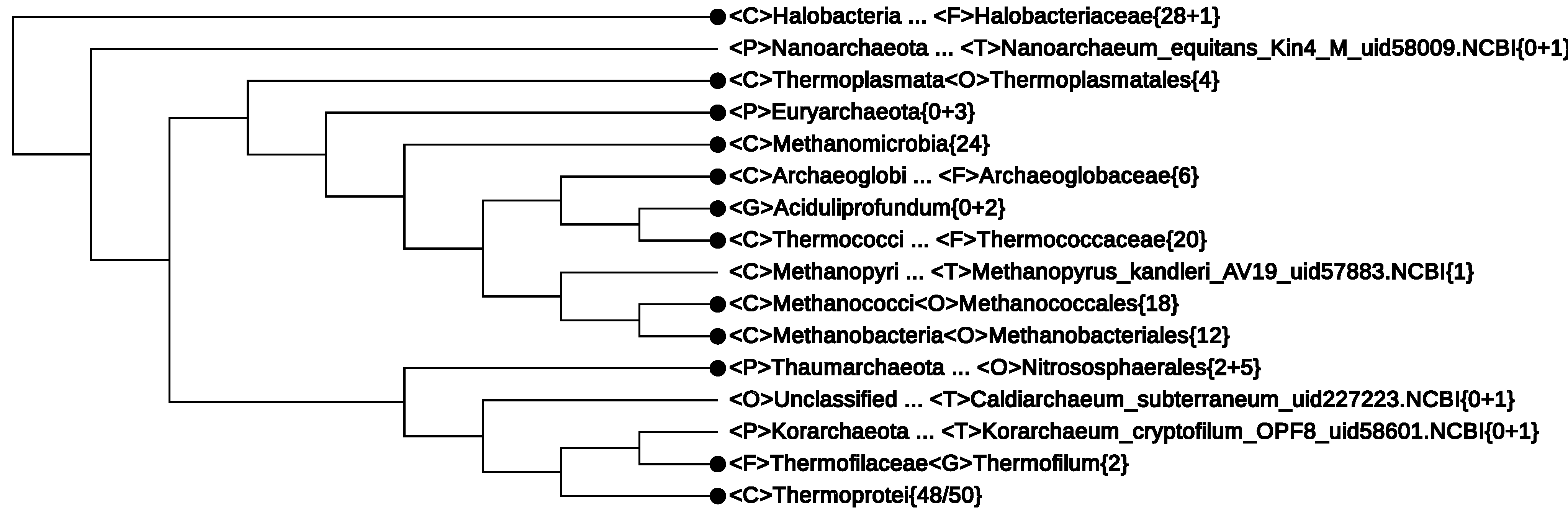
- (1)
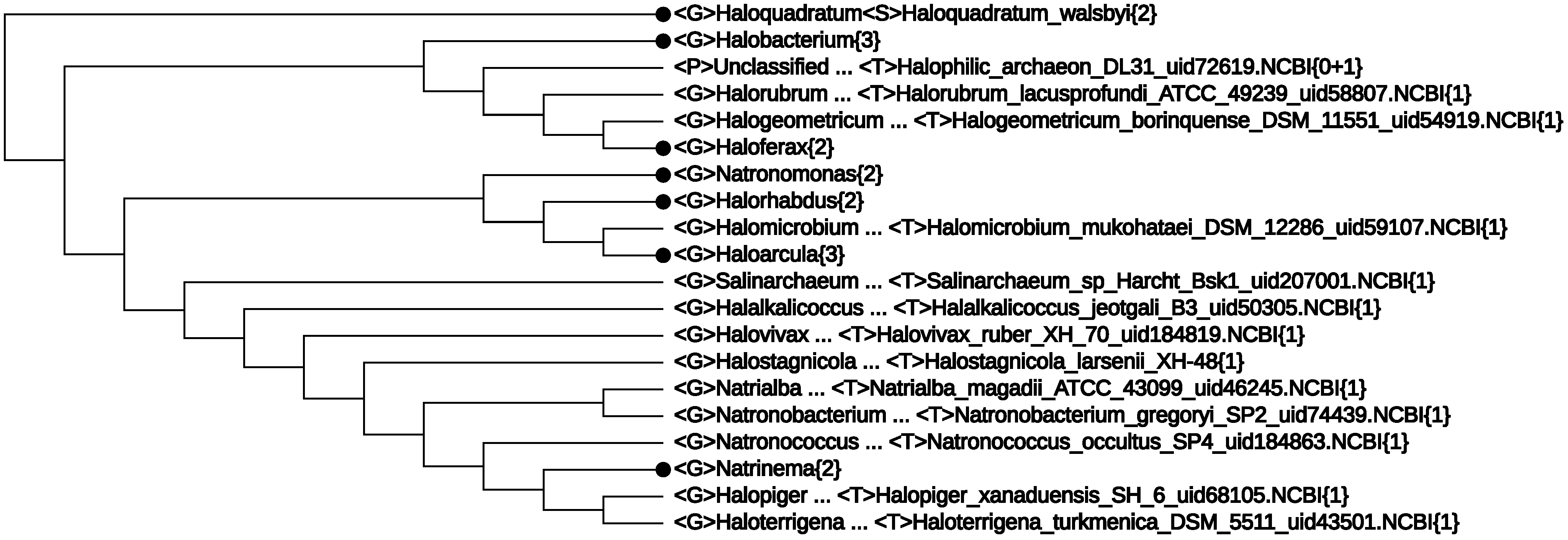
- The first line of Figure 2 <F>Halobacteriaceae{28+1} informs that among the 29 genomes, there was one without proper lineage information. In fact, it was Halophilic_archaeon_DL31_uid72619, a name not validly published and not following the basic rule for a binomen. Its NCBI lineage from phylum down to genus was “unclassified”. However, by expanding this line, the strain is seen to be located deeply inside the class Halobacteria (see Figure 4). As at present, the class consists of only one order, which, in turn, is made of one family [33], it is safe to assign this strain to a yet unspecified genus. This modification would yield a monophyletic branch, Halobacteria{29}.
- (2)
- The fourth line of Figure 2 <P>Euryarchaeota{0+3} represents a cluster obtained by collapsing three strains (not explicitly written in the figure):
- Thermoplasmatales_archaeon_BRNA1_uid195930, with NCBI lineage <C>Thermoplasmata<O>Unclassified<F>Unclassified;
- Candidatus_Methanomethylophilus_alvus_Mx1201_uid196597, with NCBI lineage <C>Unclassified<O>Unclassified<F>Unclassified,
- Methanomassiliicoccus_sp_Mx1_Issoire_uid207287, with NCBI lineage <C>Methanomicrobia<O>Unclassified<F>Unclassified.
If the NCBI lineage would be accepted, two of the above strains must violate the monophyly of the classes Thermoplasmata{4/5} and Methanomicrobia{24/25}. However, the fact that these three strains, taken together, make a monophyletic branch hint of the possibility to assign them to a yet unspecified class. This modification would restore the monophyly of the two classes Methanomicrobia{24} (Line 5 in Figure 2) and Thermoplasmata{4} (Line 3 in Figure 2), as seen in Figure 2. - (3)
- The newly proposed phylum, Thaumarchaeota, appears to be non-monophyletic, as an outlying strain, Candidatus Caldiarchaeum subterranum, was assigned to this phylum according to the NCBI taxonomy. The NCBI assignment might reflect its position in some phylogenetic tree based on concatenated proteins, e.g., Figure 2 in [52]. However, in the original paper reporting the discovery of this strain [44] and in recent 16S rRNA studies, e.g., [46], Candidatus Caldiarchaeum subterranum was proposed to make a new phylum, Aigarchaeota. CVTrees support the introduction of this new phylum. A lineage modification of Candidatus Caldiarchaeum subterranum from Thaumarchaeota to Aigarchaeota would lead to a monophyletic Thaumarchaeota.
- (4)
- The Candidatus genus, Aciduliprofundum, is considered a member of the DHEV2 (deep-sea hydrothermal vent euryarchaeotic 2) phylogenetic cluster. No taxonomic information was given in the original papers [53,54]. The NCBI taxonomy did not provide definite lineage information for this taxon at the class, order and family ranks. According to [53], the whole DHEV2 cluster was located close to Thermoplasmatales in a maximum-likelihood analysis of 16S rRNA sequences. A similar placement was seen in [52], where a Bayesian tree of the archaeal domain based on concatenation of 57 ribosomal proteins put a lonely Aciduliprofundum next to Thermoplasmata. However, in CVTrees, constructed for all K-values from three to nine, Aciduliprofundum is juxtaposed with the class Thermococci{18}. An observation in [54] that this organism shares a rare lipid structure with a few species from Thermococcales may hint to its possible association with the latter. If we temporarily presume a lineage:one might have a monophyletic class <C>Thermococci{20}.<C>Thermococci<O>Unclassified<F>Unclassified<G>Aciduliprofundum · · ·Since none of the 13 DHEV2 members listed in [53] have a sequenced genome so far, CVTree cannot tell the placement of the DHEV2 cluster as a whole for the time being. It remains an open problem whether DHEV2 is close to Thermoplasmata or to Thermococci or if a new class is needed to accommodate DHEV2.
- (5)
- The new phylum, Korarchaeota, violates the monophyly of the phylum, Crenarchaeota, by drawing to itself the family, Thermofilaceae. However, in an on-going study of ours (not published yet) using a much larger dataset, this violation no longer shows up; both Korarchaeota and Crenarchaeota restore their phylum status. Taking into account the fact that both Korarchaeota and Thermofilaceae are represented by single species for the time being, their placement certainly requires further study with broader sampling of genomes.
4.3. Phylum Distribution in Other Phylogenies
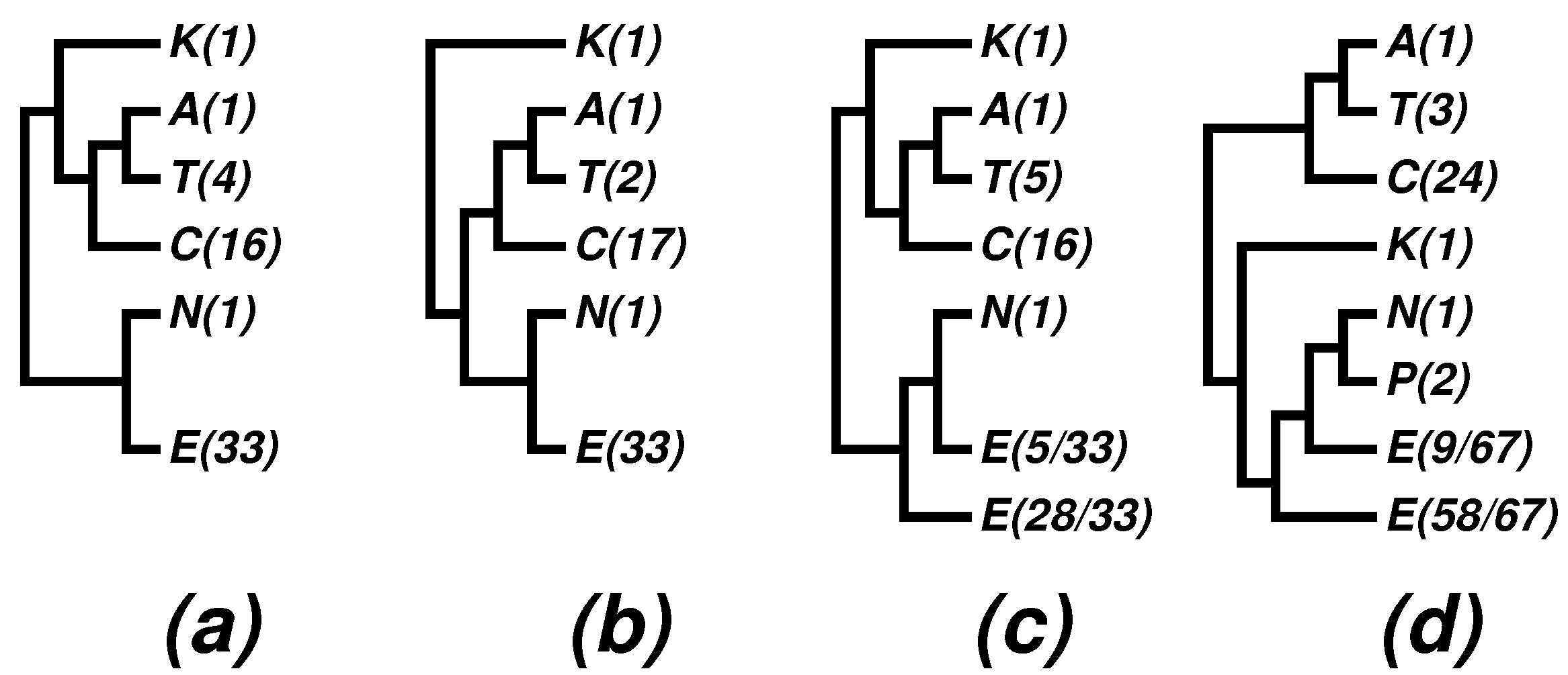
- (1)
- The newly proposed phyla, Thaumarchaeota, Korarchaeota and Aigarchaeota, are supported in many phylogenies; especially the superphylum “TACK” is supported in most phylogenies, with “TAC” being a persistent core.
- (2)
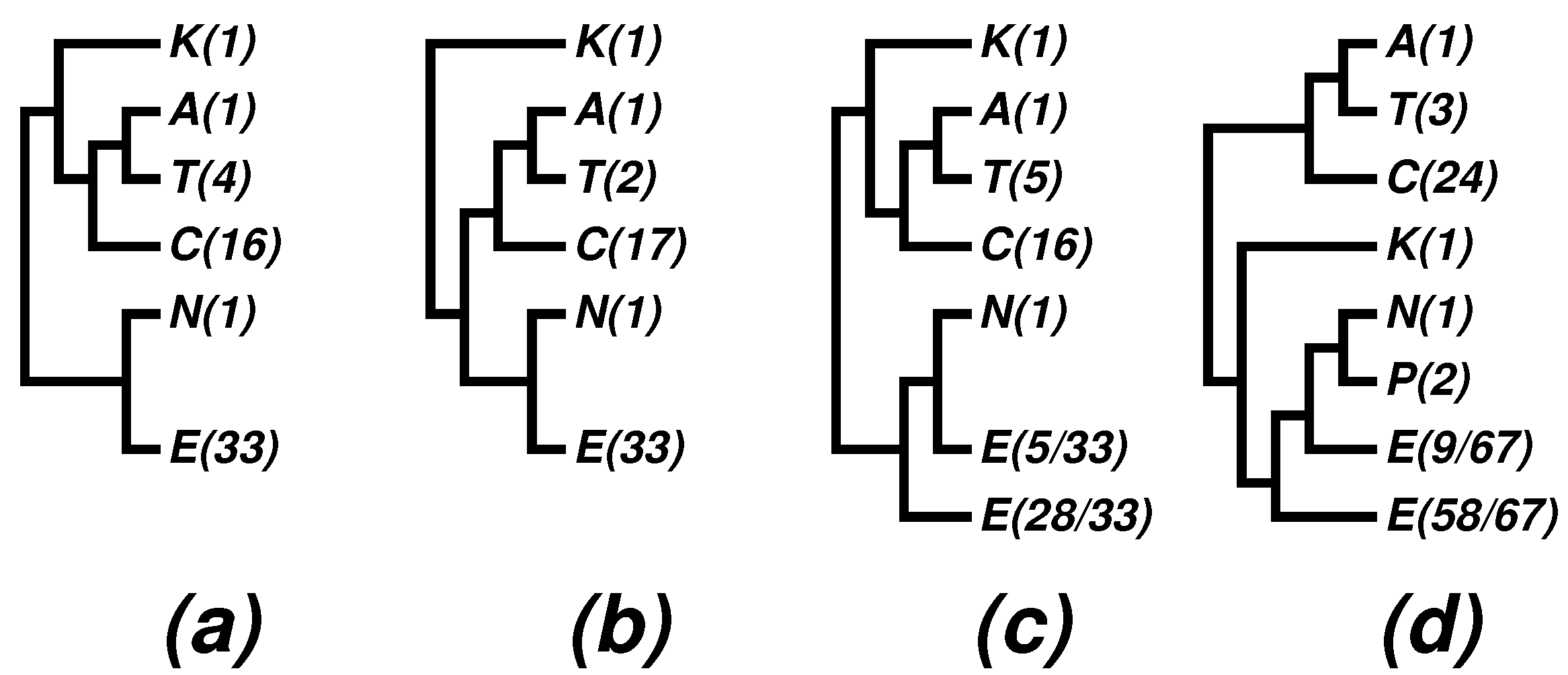
- The nano-sized archaean symbiont, Nanoarchaeum equitans, has a highly reduced genome (490,885 bp [42]). It is the only described representative of a newly proposed phylum, Nanoarchaeota, and it cuts into the otherwise monophyletic phylum, Euryarchaeota. We note that the monophyly of Euryarchaeota was also violated by Nanoarchaeum in some 16S rRNA trees; see, e.g., Figure 4 in a 2009 paper [59], as well as (c) and (d) in Figure 3. It has been known that tiny genomes of endosymbiont microbes often tend to move towards the baseline of a tree and distort the overall picture. In fact, we have suggested skipping such tiny genomes when studying bacterial phylogeny; see, e.g., [28] and a note on the home page of the CVTree web server [20]. In the present case, we may at most say that Nanoarchaeota probably makes a separate phylum, but its cutting into Euryarchaeota might be a side effect due to the tiny size of the highly-reduced genome.
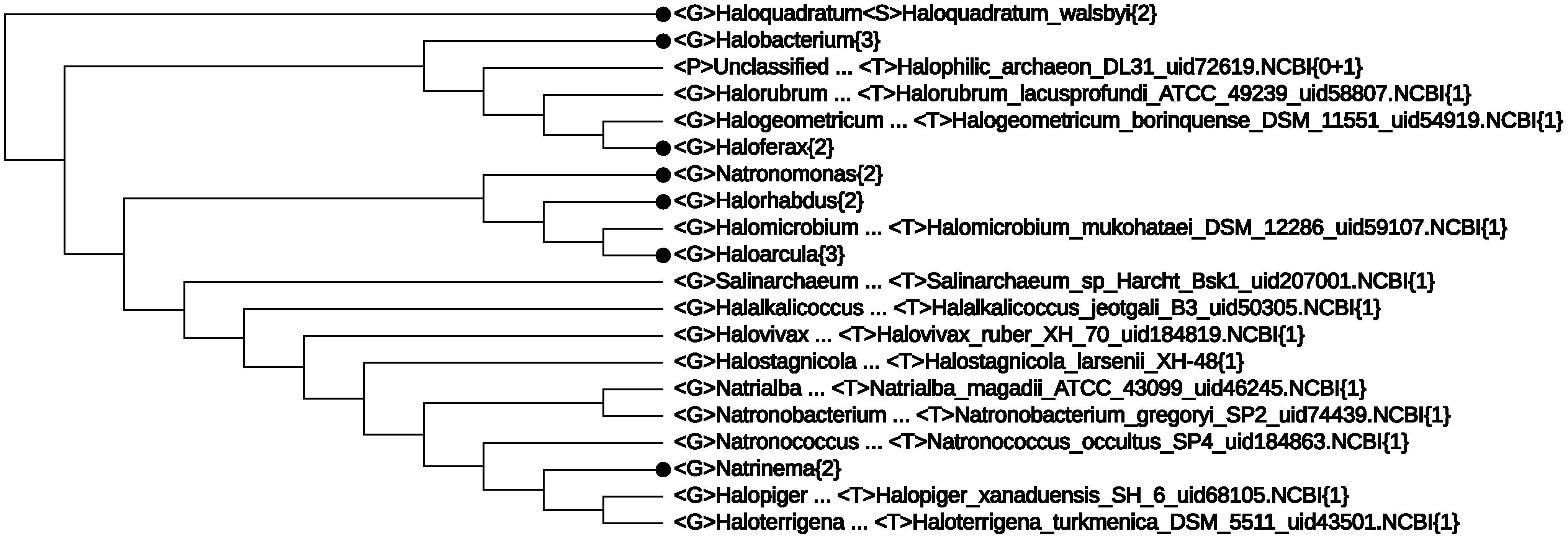
5. Conclusions
Acknowledgments
Author Contributions
Appendix: List of Genomes Used in This Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name of Strain | Proteome Size (106AA) | Accession Number |
|---|---|---|---|
| 1 | Acidianus hospitalis W1 uid66875 | 0.62 | NC_015518 |
| 2 | Acidilobus saccharovorans 345 15 uid51395 | 0.45 | NC_014374 |
| 3 | Aciduliprofundum boonei T469 uid43333 | 0.47 | NC_013926 |
| 4 | Aciduliprofundum sp. MAR08 339 uid184407 | 0.45 | NC_019942 |
| 5 | Aeropyrum camini SY1 JCM 12091 uid222311 | 0.47 | NC_022521 |
| 6 | Aeropyrum pernix K1 uid57757 | 0.49 | NC_000854 |
| 7 | Archaeoglobus fulgidus DSM 4304 uid57717 | 0.67 | NC_000917 |
| 8 | Archaeoglobus fulgidus DSM 8774 | 0.69 | CP006577 |
| 9 | Archaeoglobus profundus DSM 5631 uid43493 | 0.48 | NC_013741 |
| 10 | Archaeoglobus sulfaticallidus PM70 1 uid201033 | 0.61 | NC_021169 |
| 11 | Archaeoglobus veneficus SNP6 uid65269 | 0.56 | NC_015320 |
| 12 | Caldisphaera lagunensis DSM 15908 uid183486 | 0.44 | NC_019791 |
| 13 | Caldivirga maquilingensis IC 167 uid58711 | 0.60 | NC_009954 |
| 14 | Candidatus Caldiarchaeum subterraneum uid227223 | 0.51 | NC_022786 |
| 15 | Candidatus Korarchaeum cryptofilum OPF8 uid58601 | 0.48 | NC_010482 |
| 16 | Candidatus Methanomethylophilus alvus Mx1201 uid196597 | 0.49 | NC_020913 |
| 17 | Candidatus Nitrosopumilus koreensis AR1 uid176129 | 0.47 | NC_018655 |
| 18 | Candidatus Nitrosopumilus sp. AR2 uid176130 | 0.49 | NC_018656 |
| 19 | Candidatus Nitrososphaera evergladensis SR1 | 0.82 | CP007174 |
| 20 | Candidatus Nitrososphaera gargensis Ga9 2 uid176707 | 0.77 | NC_018719 |
| 21 | Methanomassiliicoccus sp. Mx1 Issoire uid207287 | 0.56 | NC_021353 |
| 22 | Cenarchaeum symbiosum A uid61411 | 0.62 | NC_014820 |
| 23 | Desulfurococcus fermentans DSM 16532 uid75119 | 0.40 | NC_018001 |
| 24 | Desulfurococcus kamchatkensis 1221n uid59133 | 0.40 | NC_011766 |
| 25 | Desulfurococcus mucosus DSM 2162 uid62227 | 0.39 | NC_014961 |
| 26 | Ferroglobus placidus DSM 10642 uid40863 | 0.66 | NC_013849 |
| 27 | Ferroplasma acidarmanus fer1 uid54095 | 0.57 | NC_021592 |
| 28 | Fervidicoccus fontis Kam940 uid162201 | 0.38 | NC_017461 |
| 29 | Halalkalicoccus jeotgali B3 uid50305 | 0.83 | NC_014297 |
| 30 | Haloarcula hispanica ATCC 33960 uid72475 | 1.00 | NC_0159432 |
| 31 | Haloarcula hispanica N601 uid230920 | 0.98 | NC_0230102 |
| 32 | Haloarcula marismortui ATCC 43049 uid57719 | 0.97 | NC_0063972 |
| 33 | Halobacterium salinarum R1 uid61571 | 0.60 | NC_010364 |
| 34 | Halobacterium sp. DL1 | 0.83 | CP007060 |
| 35 | Halobacterium sp. NRC 1 uid57769 | 0.59 | NC_002607 |
| 36 | Haloferax mediterranei ATCC 33500 uid167315 | 0.84 | NC_017941 |
| 37 | Haloferax volcanii DS2 uid46845 | 0.82 | NC_013967 |
| 38 | Halogeometricum borinquense DSM 11551 uid54919 | 0.82 | NC_014729 |
| 39 | Halomicrobium mukohataei DSM 12286 uid59107 | 0.90 | NC_013202 |
| 40 | Halophilic archaeon DL31 uid72619 | 0.81 | NC_015954 |
| 41 | Halopiger xanaduensis SH 6 uid68105 | 1.05 | NC_015666 |
| 42 | Haloquadratum walsbyi C23 uid162019 | 0.77 | NC_017459 |
| 43 | Haloquadratum walsbyi DSM 16790 uid58673 | 0.78 | NC_008212 |
| 44 | Halorhabdus tiamatea SARL4B uid214082 | 0.79 | NC_021921 |
| 45 | Halorhabdus utahensis DSM 12940 uid59189 | 0.91 | NC_013158 |
| 46 | Halorubrum lacusprofundi ATCC 49239 uid58807 | 0.93 | NC_0120292 |
| 47 | Halostagnicola larsenii XH-48 | 0.78 | CP007055 |
| 48 | Haloterrigena turkmenica DSM 5511 uid43501 | 1.09 | NC_013743 |
| 49 | Halovivax ruber XH 70 uid184819 | 0.91 | NC_019964 |
| 50 | Hyperthermus butylicus DSM 5456 uid57755 | 0.45 | NC_008818 |
| 51 | Ignicoccus hospitalis KIN4 I uid58365 | 0.40 | NC_009776 |
| 52 | Ignisphaera aggregans DSM 17230 uid51875 | 0.54 | NC_014471 |
| 53 | Metallosphaera cuprina Ar 4 uid66329 | 0.54 | NC_015435 |
| 54 | Metallosphaera sedula DSM 5348 uid58717 | 0.64 | NC_009440 |
| 55 | Methanobacterium formicicum strain BRM9 | 0.67 | CP006933 |
| 56 | Methanobacterium sp. AL 21 uid63623 | 0.72 | NC_015216 |
| 57 | Methanobacterium sp. MB1 complete sequence uid231690 | 0.56 | NC_023044 |
| 58 | Methanobacterium sp. SWAN 1 uid67359 | 0.66 | NC_015574 |
| 59 | Methanobrevibacter ruminantium M1 uid45857 | 0.76 | NC_013790 |
| 60 | Methanobrevibacter smithii ATCC 35061 uid58827 | 0.56 | NC_009515 |
| 61 | Methanobrevibacter sp. AbM4 uid206516 | 0.50 | NC_021355 |
| 62 | Methanocaldococcus fervens AG86 uid59347 | 0.44 | NC_013156 |
| 63 | Methanocaldococcus infernus ME uid48803 | 0.41 | NC_014122 |
| 64 | Methanocaldococcus jannaschii DSM 2661 uid57713 | 0.48 | NC_000909 |
| 65 | Methanocaldococcus sp. JH146 | 0.47 | CP009149 |
| 66 | Methanocaldococcus sp. FS406 22 uid42499 | 0.51 | NC_013887 |
| 67 | Methanocaldococcus vulcanius M7 uid41131 | 0.49 | NC_013407 |
| 68 | Methanocella arvoryzae MRE50 uid61623 | 0.89 | NC_009464 |
| 69 | Methanocella conradii HZ254 uid157911 | 0.70 | NC_017034 |
| 70 | Methanocella paludicola SANAE uid42887 | 0.86 | NC_013665 |
| 71 | Methanococcoides burtonii DSM 6242 uid58023 | 0.69 | NC_007955 |
| 72 | Methanococcus aeolicus Nankai 3 uid58823 | 0.44 | NC_009635 |
| 73 | Methanococcus maripaludis C5 uid58741 | 0.51 | NC_009135 |
| 74 | Methanococcus maripaludis C6 uid58947 | 0.51 | NC_009975 |
| 75 | Methanococcus maripaludis C7 uid58847 | 0.51 | NC_009637 |
| 76 | Methanococcus maripaludis KA1 DNA | 0.55 | AP011526 |
| 77 | Methanococcus maripaludis OS7 DNA | 0.52 | AP011528 |
| 78 | Methanococcus maripaludis S2 uid58035 | 0.49 | NC_005791 |
| 79 | Methanococcus maripaludis X1 uid70729 | 0.51 | NC_015847 |
| 80 | Methanococcus vannielii SB uid58767 | 0.49 | NC_009634 |
| 81 | Methanococcus voltae A3 uid49529 | 0.51 | NC_014222 |
| 82 | Methanocorpusculum labreanum Z uid58785 | 0.52 | NC_008942 |
| 83 | Methanoculleus bourgensis MS2T uid171377 | 0.77 | NC_018227 |
| 84 | Methanoculleus marisnigri JR1 uid58561 | 0.72 | NC_009051 |
| 85 | Methanohalobium evestigatum Z 7303 uid49857 | 0.63 | NC_014253 |
| 86 | Methanohalophilus mahii DSM 5219 uid47313 | 0.59 | NC_014002 |
| 87 | Methanolobus psychrophilus R15 uid177925 | 0.87 | NC_018876 |
| 88 | Methanomethylovorans hollandica DSM 15978 uid184864 | 0.69 | NC_019977 |
| 89 | Methanoplanus petrolearius DSM 11571 uid52695 | 0.83 | NC_014507 |
| 90 | Methanopyrus kandleri AV19 uid57883 | 0.50 | NC_003551 |
| 91 | Methanoregula boonei 6A8 uid58815 | 0.73 | NC_009712 |
| 92 | Methanoregula formicicum SMSP uid184406 | 0.81 | NC_019943 |
| 93 | Methanosaeta concilii GP6 uid66207 | 0.84 | NC_015416 |
| 94 | Methanosaeta harundinacea 6Ac uid81199 | 0.73 | NC_017527 |
| 95 | Methanosaeta thermophila PT uid58469 | 0.51 | NC_008553 |
| 96 | Methanosalsum zhilinae DSM 4017 uid68249 | 0.61 | NC_015676 |
| 97 | Methanosarcina acetivorans C2A uid57879 | 1.42 | NC_003552 |
| 98 | Methanosarcina barkeri str Fusaro uid57715 | 1.12 | NC_007355 |
| 99 | Methanosarcina mazei Go1 uid57893 | 1.02 | NC_003901 |
| 100 | Methanosarcina mazei Tuc01 uid190185 | 0.82 | NC_020389 |
| 101 | Methanosphaera stadtmanae DSM 3091 uid58407 | 0.49 | NC_007681 |
| 102 | Methanosphaerula palustris E1 9c uid59193 | 0.82 | NC_011832 |
| 103 | Methanospirillum hungatei JF 1 uid58181 | 1.01 | NC_007796 |
| 104 | Methanothermobacter marburgensis str Marburg uid51637 | 0.49 | NC_014408 |
| 105 | Methanothermobacter thermautotrophicus str Delta H uid57877 | 0.53 | NC_000916 |
| 106 | Methanothermobacter thermautotrophicus CaT2 DNA | 0.51 | AP011952 |
| 107 | Methanothermococcus okinawensis IH1 uid51535 | 0.45 | NC_015636 |
| 108 | Methanothermus fervidus DSM 2088 uid60167 | 0.38 | NC_014658 |
| 109 | Methanotorris igneus Kol 5 uid67321 | 0.51 | NC_015562 |
| 110 | Nanoarchaeum equitans Kin4 M uid58009 | 0.15 | NC_005213 |
| 111 | Natrialba magadii ATCC 43099 uid46245 | 1.05 | NC_013922 |
| 112 | Natrinema pellirubrum DSM 15624 uid74437 | 1.06 | NC_019962 |
| 113 | Natrinema sp. J7 2 uid171337 | 1.05 | NC_018224 |
| 114 | Natronobacterium gregoryi SP2 uid74439 | 1.04 | NC_019792 |
| 115 | Natronococcus occultus SP4 uid184863 | 1.12 | NC_019974 |
| 116 | Natronomonas moolapensis 8 8 11 uid190182 | 0.82 | NC_020388 |
| 117 | Natronomonas pharaonis DSM 2160 uid58435 | 0.78 | NC_007426 |
| 118 | Nitrosopumilus maritimus SCM1 uid58903 | 0.49 | NC_010085 |
| 119 | Nitrososphaera viennensis EN76 | 0.73 | CP007536 |
| 120 | Palaeococcus pacificus DY20341 | 0.56 | CP006019 |
| 121 | Picrophilus torridus DSM 9790 uid58041 | 0.47 | NC_005877 |
| 122 | Pyrobaculum aerophilum str IM2 uid57727 | 0.66 | NC_003364 |
| 123 | Pyrobaculum arsenaticum DSM 13514 uid58409 | 0.61 | NC_009376 |
| 124 | Pyrobaculum calidifontis JCM 11548 uid58787 | 0.61 | NC_009073 |
| 125 | Pyrobaculum islandicum DSM 4184 uid58635 | 0.53 | NC_008701 |
| 126 | Pyrobaculum neutrophilum V24Sta uid58421 | 0.53 | NC_010525 |
| 127 | Pyrobaculum oguniense TE7 uid84411 | 0.71 | NC_016885 |
| 128 | Pyrobaculum sp. 1860 uid82379 | 0.73 | NC_016645 |
| 129 | Pyrococcus abyssi GE5 uid62903 | 0.54 | NC_000868 |
| 130 | Pyrococcus furiosus COM1 uid169620 | 0.57 | NC_018092 |
| 131 | Pyrococcus furiosus DSM 3638 uid57873 | 0.59 | NC_003413 |
| 132 | Pyrococcus horikoshii OT3 uid57753 | 0.55 | NC_000961 |
| 133 | Pyrococcus sp. NA2 uid66551 | 0.57 | NC_015474 |
| 134 | Pyrococcus sp. ST04 uid167261 | 0.52 | NC_017946 |
| 135 | Pyrococcus yayanosii CH1 uid68281 | 0.51 | NC_015680 |
| 136 | Pyrolobus fumarii 1A uid73415 | 0.54 | NC_015931 |
| 137 | Salinarchaeum sp. Harcht Bsk1 uid207001 | 0.91 | NC_021313 |
| 138 | Staphylothermus hellenicus DSM 12710 uid45893 | 0.46 | NC_014205 |
| 139 | Staphylothermus marinus F1 uid58719 | 0.46 | NC_009033 |
| 140 | Sulfolobus acidocaldarius DSM 639 uid58379 | 0.63 | NC_007181 |
| 141 | Sulfolobus acidocaldarius N8 uid189027 | 0.62 | NC_020246 |
| 142 | Sulfolobus acidocaldarius Ron12 I uid189028 | 0.64 | NC_020247 |
| 143 | Sulfolobus acidocaldarius SUSAZ uid232254 | 0.59 | NC_023069 |
| 144 | Sulfolobus islandicus HVE10 4 uid162067 | 0.76 | NC_017275 |
| 145 | Sulfolobus islandicus L D 8 5 uid43679 | 0.77 | NC_013769 |
| 146 | Sulfolobus islandicus L S 2 15 uid58871 | 0.76 | NC_012589 |
| 147 | Sulfolobus islandicus LAL14 1 uid197216 | 0.71 | NC_021058 |
| 148 | Sulfolobus islandicus M 14 25 uid58849 | 0.74 | NC_012588 |
| 149 | Sulfolobus islandicus M 16 27 uid58851 | 0.76 | NC_012632 |
| 150 | Sulfolobus islandicus M 16 4 uid58841 | 0.75 | NC_012726 |
| 151 | Sulfolobus islandicus REY15A uid162071 | 0.72 | NC_017276 |
| 152 | Sulfolobus islandicus Y G 57 14 uid58923 | 0.78 | NC_012622 |
| 153 | Sulfolobus islandicus Y N 15 51 uid58825 | 0.77 | NC_012623 |
| 154 | Sulfolobus solfataricus 98 2 uid167998 | 0.72 | NC_017274 |
| 155 | Sulfolobus solfataricus P2 uid57721 | 0.84 | NC_002754 |
| 156 | Sulfolobus tokodaii str 7 uid57807 | 0.76 | NC_003106 |
| 157 | Thermococcus barophilus MP uid54733 | 0.62 | NC_014804 |
| 158 | Thermococcus eurythermalis strain A501 | 0.60 | CP008887 |
| 159 | Thermococcus gammatolerans EJ3 uid59389 | 0.64 | NC_012804 |
| 160 | Thermococcus kodakarensis KOD1 uid58225 | 0.64 | NC_006624 |
| 161 | Thermococcus litoralis DSM 5473 uid82997 | 0.67 | NC_022084 |
| 162 | Thermococcus nautili strain 30 1 | 0.61 | CP007264 |
| 163 | Thermococcus onnurineus NA1 uid59043 | 0.56 | NC_011529 |
| 164 | Thermococcus sibiricus MM 739 uid59399 | 0.55 | NC_012883 |
| 165 | Thermococcus sp. 4557 uid70841 | 0.61 | NC_015865 |
| 166 | Thermococcus sp. AM4 uid54735 | 0.63 | NC_016051 |
| 167 | Thermococcus sp. CL1 uid168259 | 0.58 | NC_018015 |
| 168 | Thermococcus sp. ES1 | 0.58 | CP006965 |
| 169 | Thermofilum pendens Hrk 5 uid58563 | 0.54 | NC_008698 |
| 170 | Thermofilum sp. 1910b uid215374 | 0.52 | NC_022093 |
| 171 | Thermogladius cellulolyticus 1633 uid167488 | 0.41 | NC_017954 |
| 172 | Thermoplasma acidophilum DSM 1728 uid61573 | 0.45 | NC_002578 |
| 173 | Thermoplasma volcanium GSS1 uid57751 | 0.45 | NC_002689 |
| 174 | Thermoplasmatales archaeon BRNA1 uid195930 | 0.44 | NC_020892 |
| 175 | Thermoproteus tenax Kra 1 uid74443 | 0.55 | NC_016070 |
| 176 | Thermoproteus uzoniensis 768 20 uid65089 | 0.59 | NC_015315 |
| 177 | Thermosphaera aggregans DSM 11486 uid48993 | 0.40 | NC_014160 |
| 178 | Vulcanisaeta distributa DSM 14429 uid52827 | 0.71 | NC_014537 |
| 179 | Vulcanisaeta moutnovskia 768 28 uid63631 | 0.67 | NC_015151 |
Conflicts of Interest
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.E.; Magrum, L.J.; Balch, W.E.; Wolfe, R.S.; Woese, C.R. Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proc. Natl. Acad. Sci. USA 1977, 74, 4537–4541. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.E.; Pechman, K.R.; Woese, C.R. Comarative cataloging of 16S ribosomal ribonucleic acid: Molecular approach to procaryotic systematics. Int. J. Syst. Bacteriol. 1977, 27, 44–57. [Google Scholar] [CrossRef]
- The Bergey’s Manual Trust. In Bergey’s Manual of Systematic bacteriology, 2nd ed.; Springer: New York, NY, USA; Volumes 1∼5, pp. 2001–2012.
- Konstantinidis, K.T.; Tiedje, J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar]
- Qi, J.; Wang, B.; Hao, B. Whole genome prokaryote phylogeny without sequence alignment: A K-string composition approach. J. Mol. Evol. 2004, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Qi, J. Prokaryote phylogeny without sequence alignment: From avoidance signature to composition distance. J. Bioinf. Comput. Biol. 2004, 2. [Google Scholar] [CrossRef]
- Gao, L.; Qi, J.; Sun, J.; Hao, B. Prokaryote phylogeny meets taxonomy: An exhaustive comparison of composition vector trees with systematic bacteriology. Sci. China Life Sci. 2007, 50, 587–599. [Google Scholar] [CrossRef]
- Li, Q.; Xu, Z.; Hao, B. Composition vector approach to whole-genome-based prokaryotic phylogeny: Success and foundations. J. Biotech. 2010, 149, 115–119. [Google Scholar] [CrossRef]
- Zuo, G.; Xu, Z.; Yu, H.; Hao, B. Jackknife and bootstrap tests of the composition vector trees. Genomics Proteomics Bioinform. 2010, 8, 262–267. [Google Scholar] [CrossRef]
- Hao, B. CVTrees support the Bergey’s systematics and provide high resolution at species level and below. Bull. BISMiS 2011, 2, 189–196. [Google Scholar]
- Chan, P.P.; Cozen, A.E.; Lowe, T.M. Reclassification of Thermoproteus neutrophilus Stetter and Zillig 1989 as Pyrobaculum neutrophilum comb. nov., based on phylogenetic analysis. Int. J. Syst. Evol. Microbiol. 2013, 63, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int. J. Syst. Evol. Microbiol. 2002, 52, 7–76. [Google Scholar] [PubMed]
- Lapage, S.P.; Sneath, P.H.A.; Lessel, E.F.; Skerman, V.B.D.; Seeliger, H.P.R.; Clark, W.A. International Code of Nomenclature of Bacteria: Bacteriological Code 1990; ASM Press: Washington, DC, USA, 1992. [Google Scholar]
- De Vos, P.; Trüper, H.G. Judicial Commission of the International Committee on Systematic Bacteriology. Int. J. Syst. Evol. Microbiol. 2000, 50, 2239–2244. [Google Scholar] [CrossRef]
- The GOLD (Genomes On Line Database) site. Available online: https://gold.jgi-psf.org (accessed on 12 February 2015).
- PATRIC (Pathosystems Resource Integration Center). Available online: http://particbrc.org/portal/portal/patric/Genomes (accessed on 12 February 2015).
- The NCBI FTP site. Available online: ftp://ftp.ncbi.nih.gov/genomes/Bacteria/ (accessed on 27 February 2015).
- Xu, Z.; Hao, B. CVTree update: A newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res. 2009, 37, W174–W178. [Google Scholar] [CrossRef] [PubMed]
- The much improved CVTree3 Web Server. Available online: http://tlife.fudan.edu.cn/cvtree3/ (accessed on 25 February 2015).
- The EBI Archaea genome list. Available online: http://www.ebi.ac.uk/genomes/archaea.html (accessed on 15 February 2015).
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Pess: Cambridge, UK, 1985. [Google Scholar]
- Woese, C. The universal ancestor. Proc. Natl. Acad, Sci. USA 1998, 95, 6854–6859. [Google Scholar] [CrossRef]
- Wagner, A; de la Chaus, N. Distant horizontal gene transfer is rare for multiple families of prokaryotic insertion sequences. Mol. Genet. Genomics 2008, 280, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Luo, H.; Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004, 32, W45–W47. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Li, Q.; Hao, B. On K-peptide length in composition vector phylogeny of prokaryotes. Comput. Biol. Chem. 2014, 53, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hao, B. Whole-genome based prokaryotic branches in the Tree of Life. In Darwin’s Heritage Today: Proceedings of the Darwin 200 Beijing International Conference; Long, M., Gu, H., Zhou, Z., Eds.; High Education Press: Beijing, China, 2010; pp. 102–113. [Google Scholar]
- Garrity, G.M.; Holt, J.G. Taxonomic Outline of the Archaea and Bacteria. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Boone, D.R., Castenholz, R.W., Eds.; Springer: New York, NY, USA, 2001; Volume 1, pp. 155–156. [Google Scholar]
- Parte, A.C. LPSN—list of prokaryotic names with standing in Nomenclature. Nucleic Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed]
- Prokofeva, M.I.; Kostrikina, N.A.; Kolganova, T.V.; Tourova, T.P.; Lysenko, A.M.; Lebedinsky, A.V.; Bonch-Osmolovskaya1, F.A. Isolation of the anaerobic thermoacidophilic crenarchaeote Acidilobus saccharovorans sp. nov. and proposal of Acidilobales ord. nov., including Acidilobaceae fam. nov. and Caldisphaeraceae fam. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 3116–3122. [Google Scholar] [CrossRef] [PubMed]
- Perevalova, A.A.; Bidzhieva, S.K.; Kublanov, I.V.; Hinrichs, K.-U.; Liu, X.L.; Mardanov, A.V.; Lebedinsky, A.V.; Bonch-Osmolovskaya, E.A. Fervidicoccus fontis gen. nov., sp. nov., an anaerobic, thermophilic crenarchaeote from terrestrial hot springs, and proposal of Fervidicoccaceae fam. nov. and Fervidicoccales ord. nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Garrity, G.M.; Bell, J.A.; Lilburn, T.G. The Revised Roadmap to the Manual. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Springer: New York, NY, USA, 2005; Volume 2, pp. 159–187. [Google Scholar]
- Sakai, S.; Imachi, H.; Hanada, S.; Ohashi, A.; Harada1, H.; Kamagata, Y. Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage “Rice Cluster I”, and proposal of the new archaeal order Methanocellales ord. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Naushad, S.; Baker, S. Phylogenomic analyses and molecular signatures for the class Halobacteria and its two major clades: A proposal for division of the class Halobacteria into an emended order Halobacteriales and two new orders, Haloferacales ord. nov. and Natrialbales ord. nov. Int. J. Syst. Evol. Microbiol. 2014. [CrossRef]
- Barns, S.M.; Delwiche, C.F.; Palmer, J.D.; Pace, N.R. Perspectives on archaeal diversity, thermophyly and monophyly from environmental rRNA sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 9188–9193. [Google Scholar] [CrossRef] [PubMed]
- Auchtung, T.A.; Shyndriayeva, G.; Cavanaugh, C.M. 16S rRNA phylogenetic analysis and quantification of Koarchaeota indigenous to the hot springs of Kamchatka, Russia. Extremophiles 2011, 15, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Brochier-Armanet, C.; Boussau, B.; Gribaldo, S.; Forterre, P. Mesophilic crenarchaeota: Proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 2008, 6, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Shami, A. Molecular signatures for the Crenarchaeota and Thaumarchaeota. Antonie van Leeuwenhoek 2011, 99, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Pester, M.; Schleper, C.; Wagner, M. The Thaumarchaeota: An emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 2011, 14, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.; Hohn, M.J.; Rachel, R.; Fuchs, T.; Wimmer, V.C.; Stetter, K.O. A new phylum of Archaea represented by a nano-sized hyperthermophilic symbiont. Nature 2002, 417, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.; Hohn, M.J.; Ahel, I.; Graham, D.E.; Adams, M.D.; Barnstead, M.; Beeson, K.Y.; Bibbs, L.; Bolanos, R.; Keller, M.; et al. The genome of Nanoarchaeum equitan: Insights into early archaeal evolution and derived parasitism. Proc. Natl. Aad. Sci. USA 2003, 100, 12984–12988. [Google Scholar] [CrossRef]
- Clingenpeel, S.; Kan, J.; Macur, R.E.; Woyke, T.; Lavalvo, D.; Carley, J.; Inskeep, W.P.; Nealson, K.; McDermott, T. Yellowstone Lake Nanoarchaeota. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Nunoura1, T.; Takaki, Y.; Kakuta, J.; Nishi, S.; Sugahara, J.; Kazama, H.; Chee, G.-J.; Hattori, M.; Kanai, A.; Atomi, H.; et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 2011, 39, 3204–3223. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Comolli, L.R.; Dick, G.J.; Hauser, L.J.; Haytt, D.; Dill, B.J.; Land, M.L.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. Enegmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xu, J.; Qin, D.; He, Y.; Xiao, X.; Wang, F. Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 2014, 8, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Richter, M.; Peplies, J.; Euzéby, J.; Amann, R.; Schleifer, K.-H.; Ludwig, W.; Glöckner, F.O.; Roselló-Móra, R. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all se-quenced type strains. Syst. Appl. Microbiol. 2008, 31, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Ludwig, W.; Euzéby, J.; Amann, R.; Schleifer, K.-H.; Glöckner, F.O.; Rossweló-Móra, R. Update of the All-Species Living Tree project based on 16S and 23S rRNA sequence analysis. Syst. Appl. Microbiol. 2010, 33, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Wegener-Parfrey, L.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- LTPs115 web site. Available online: http://www.silva-arb.de/projects/livibg-tree/ (accessed on 25 November 2014).
- LVTree Viewer. Available online: http://tlife.fudan.edu.cn/lvtree/ (accessed on 25 November 2014).
- Brochier-Armanet, C.; Forterre, P.; Gribaldo, S. Phylogeny and evolution of the Archaea: One hundred genomes later. Curr. Opin. Microbiol. 2011, 14, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Reysenbach, A.-L.; Liu, Y.; Banta, A.B.; Beveridge, T.J.; Kirshtein, J.D.; Schouten, S.; Tivey, M.K.; von Damm, K.L.; Voytek, M.A. A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature 2006, 422, 444–447. [Google Scholar] [CrossRef]
- Schouten, S.; Baas, M.; Hopmans, E.C.; Reysenbach, A.-L.; Sinninghe Damste, J.S. Tetraether membrane lipids of Candidatus “Aciduliprofundum boonei”, a cultivated obligate thermoacidophilic euryarchaeote from deep-sea hydrothermal vents. Extremophiles 2008, 12, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Guy, L.; Ettema, T.J.G. The archaeal “TACK” superphylum and the origin of eukaryotes. Trends Micrbiol. 2011, 19, 580–587. [Google Scholar] [CrossRef]
- Sun, J.; Xu, Z.; Hao, B. Whole-genome based Archaea phylogeny and taxonomy: A composition vector approach. Chin. Sci. Bull. 2010, 55, 2323–2328. [Google Scholar] [CrossRef]
- Daubin, V.; Gouy, M.; Perriére, G. Bacterial molecular phylogeny using supertree approach. Genome Inform. 2001, 12, 155–164. [Google Scholar] [PubMed]
- Wolf, Y.I.; Rogiozin, I.B.; Grishin, N.V.; Tatusov, R.L.; Koonin, E.V. Genome tree constructed using five different approaches suggest new major bacterial clades. BMC Evol. Biol. 2001, 1. [Google Scholar] [CrossRef]
- Gribaldo, S.; Brochier, C. Phylogeny of prokaryotes: Does it exist and why should we care? Res. Microbiol. 2009, 160, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Hao, B.; Staley, J.R. Geographic divergence of “Sulfolobus islandicus” strains assessed by genomic analyses including electronic DNA hybridization confirms they are geovars. Antonie van Leeuwenoek 2014, 105, 431–435. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, G.; Xu, Z.; Hao, B. Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis. Life 2015, 5, 949-968. https://doi.org/10.3390/life5010949
Zuo G, Xu Z, Hao B. Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis. Life. 2015; 5(1):949-968. https://doi.org/10.3390/life5010949
Chicago/Turabian StyleZuo, Guanghong, Zhao Xu, and Bailin Hao. 2015. "Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis" Life 5, no. 1: 949-968. https://doi.org/10.3390/life5010949
APA StyleZuo, G., Xu, Z., & Hao, B. (2015). Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis. Life, 5(1), 949-968. https://doi.org/10.3390/life5010949