Viruses of Haloarchaea

Abstract
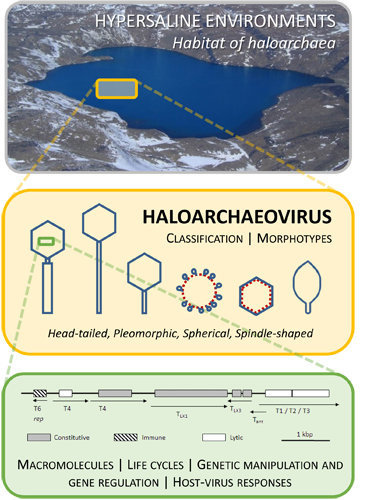
:
1. Introduction
2. Classification
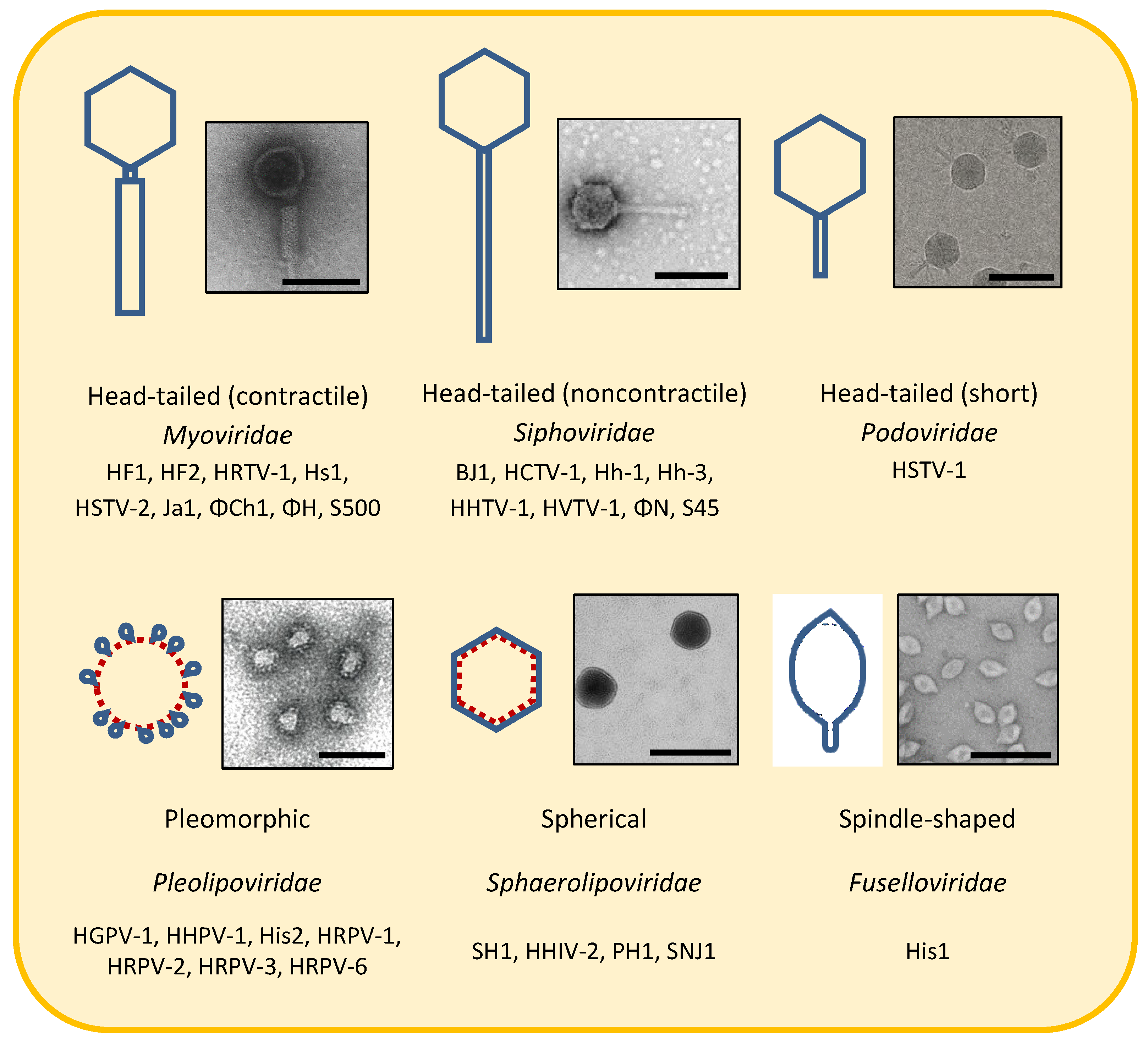
Viral Lineages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus 1 | Host 2 | Mode of Infection 3 | Source | Morphology 4 | Size/nm | Genome Type | Genome Size/kb | G + C mol% | pI | References |
|---|---|---|---|---|---|---|---|---|---|---|
| HATV-1 | Haloarcula sp. | Virulent | Saltern, Thailand | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HATV-2 | Har. sp., Halorubrum. sp. | Virulent | Saltern, Israel | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HF1 | Halobacterium salinarum, Haloferax volcanii | Persistent | Saltern, Australia | Head-tailed (contractile) | Head 67.8 ± 3, Tail 90 ± 2 | Linear dsDNA | 75.9 | 55.8 | <5 | [25,45] |
| HF2 | Halorubrum coriense, Halorubrum saccharovorum | Persistent | Saltern, Australia | Head-tailed (contractile) | Head 58, Tail 94 | Linear dsDNA | 77.7 | 55.8 | <5 | [25,45,46] |
| HGTV-1 | Halogranum sp. | Virulent | Saltern, Thailand | Head-tailed (contractile) | - | Linear dsDNA | 143.9 | 50.4 | - | [2,47] |
| HJTV-1 | Haloarcula japonica | Virulent | Saltern, Italy | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HJTV-2 | Har. Japonica | Virulent | Saltern, Thailand | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-1 | Hrr. sp. | - | Saltern, Italy | Head-tailed (contractile) | Head 50, Tail 87 | - | - | - | - | [48] |
| HRTV-2 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-3 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-5 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (contractile) | - | Linear dsDNA | 76.1 | 56.4 | - | [2,47] |
| HRTV-6 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-7 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (contractile) | - | Linear dsDNA | 69.0 | 59.6 | - | [2,47] |
| HRTV-8 | Hrr. sp. | Virulent | Saltern, Thailand | Head-tailed (contractile) | - | Linear dsDNA | 74.5 | 57.1 | - | [2,47] |
| HRTV-9 | Hrr. sp. | Virulent | Saltern, Israel | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-10 | Hrr. sp. | Virulent | Saltern, Israel | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-11 | Hrr. sp. | Virulent | Saltern, Israel | Head-tailed (contractile) | - | - | - | - | - | [2] |
| HRTV-12 | Hrr. sp. | Virulent | Saltern, Spain | Head-tailed (contractile) | - | - | - | - | - | [2] |
| Hs1 | Hbt. Salinarum | Persistent | Hbt. salinarum cultures from salted codfish | Head-tailed (contractile) | Head 50, Tail 120 | - | - | - | - | [14,49] |
| HSTV-2 | Halorubrum. Sodomense | Virulent | Saltern, Israel | Head-tailed (contractile) | Head 60, Tail 101 ± 5 | Linear dsDNA | 68.2 | 60.0 | - | [2,50] |
| HSTV-3 | Hrr. sodomense | Virulent | Saltern, Israel | Head-tailed (contractile) | - | - | - | - | - | [2] |
| Ja1 | Hbt. Salinarum | Virulent | Salt ponds, Jamaica | Head-tailed (contractile) | Head 90, Tail 150 | - | - | - | - | [5] |
| ΦCh1 | Natrialba magadii | Temperate | Nab. magadii culture | Head-tailed (contractile) | Head 70, Tail 130 | Linear dsDNA | 58.5 | 61.9 | <5.2 | [51,52,53] |
| ΦH | Hbt. Salinarum | Temperate | Hbt. salinarum culture | Head-tailed (contractile) | Head 64, Tail 170 | Linear dsDNA | 59.0 | 65.0 | - | [54,55,56,57,58,59,60,61] |
| S5100 | Hbt. salinarum | Persistent | Salt ponds, Jamaica | Head-tailed (contractile) | - | dsDNA | - | - | - | [62] |
| BJ1 | Hrr. sp. | Temperate | Salt lake, Mongolia | Head-tailed (non-contractile) | Head 56, Tail 71 | Linear dsDNA | 42.3 | 64.0 | <5 | [63] |
| HCTV-1 | Haloarcula californiae | Virulent | Saltern, Italy | Head-tailed (non-contractile) | Head 63, Tail 80 | Linear dsDNA | 103.2 | 57.0 | - | [47,48] |
| HCTV-2 | Har. californiae | Virulent | Saltern, Thailand | Head-tailed (non-contractile) | - | Linear dsDNA | 54.3 | 68.1 | - | [2,47] |
| HCTV-5 | Har. californiae | Virulent | Saltern, Thailand | Head-tailed (non-contractile) | - | Linear dsDNA | 102.1 | 57.6 | - | [2,47] |
| Hh-1 | Hbt. salinarum | Persistent | Anchovy sauce, Philippines | Head-tailed (non-contractile) | Head 60, Tail 100 | dsDNA | 32.7 | 67.1 | - | [64,65] |
| Hh-3 | Hbt. salinarum | Persistent | Anchovy sauce, Philippines | Head-tailed (non-contractile) | Head 75, Tail 50 | dsDNA | 29.4 | 62.2 | - | [64,65] |
| HHTV-1 | Haloarcula hispanica | Virulent | Saltern, Italy | Head-tailed (non-contractile) | Head 50, Tail 110 | Linear dsDNA | 49.1 | 56.5 | - | [47,48] |
| HHTV-2 | Har. hispanica | Virulent | Saltern, Thailand | Head-tailed (non-contractile) | - | Linear dsDNA | 52.6 | 66.6 | - | [2,47] |
| HRTV-4 | Hrr. sp. | Virulent | Saltern, Italy | Head-tailed (non-contractile) | - | Linear dsDNA | 35.7 | 59.5 | - | [2,47] |
| HVTV-1 | Haloarcula vallismortis | Virulent | Saltern, Thailand | Head-tailed (non-contractile) | Head 70, Tail 73 ± 5 | Linear dsDNA | 101.7 | 58.0 | - | [2,50] |
| ΦN | Hbt. salinarum | Virulent | Hbt. salinarum NRL/JW cultures | Head-tailed (non-contractile) | Head 55, Tail 85 | Linear dsDNA | 56.0 | 70.0 | - | [66] |
| S45 | Hbt. salinarum | Virulent | Salt ponds, Jamaica | Head-tailed (non-contractile) | Head 40, Tail 70 | dsDNA | - | - | - | [67] |
| HSTV-1 | Haloarcula sinaiiensis, Har. vallismortis | Virulent | Saltern, Italy | Head-tailed (short) | Head 60, Tail 40 | Circular dsDNA | 32.2 | 60.0 | - | [2,38] |
| EHP-2 | Putative Haloquadratum walsbyi | - | Saltern, Spain | Putative head-tailed | - | Linear dsDNA | 27.2 | 43.9 | - | [12] |
| EPH-11 | Putative Halorubrum lacusprofundi | - | Saltern, Spain | Putative head-tailed | - | Linear dsDNA | 33.5 | 58.5 | - | [12] |
| HGPV-1 | Halogeometricum sp. | Persistent | Saltern, Spain | Pleomorphic | 55.5 ± 5.2 | Circular dsDNA | 9.7 | 61.6 | - | [2,26,68] |
| HHPV-1 | Har. hispanica | Persistent | Saltern, Italy | Pleomorphic | 51.7 ± 4.0 | Circular dsDNA | 8.1 | 55.8 | - | [26,35,48,69] |
| His2 | Har. hispanica | Persistent | Salt lake, Australia | Pleomorphic | 70.6 ± 3.6 | Linear dsDNA | 16.1 | 39.0–40.0 | <7 | [24,26,70] |
| HRPV-1 | Hrr. sp. | Persistent | Saltern, Italy | Pleomorphic | 41.1 ± 2.2 | Circular ssDNA | 7.0 | 54.2 | - | [26,69] |
| HRPV-2 | Hrr. sp. | Persistent | Saltern, Thailand | Pleomorphic | 54.0 ± 4.3 | ssDNA | 10.7 | 63.7 | - | [2,26] |
| HRPV-3 | Hrr. sp. | Persistent | Artificial salt pond, Israel | Pleomorphic | 67.2 ± 5.2 | Circular dsDNA | 8.8 | 58.3 | - | [2,26,68] |
| HRPV-6 | Hrr. sp. | Persistent | Saltern, Thailand | Pleomorphic | 48.5 ± 2.7 | Circular ssDNA | 8.5 | 62.7 | - | [26,68] |
| SH1 | Har. hispanica | Persistent | Salt lake, Australia | Spherical | 70 | Linear dsDNA | 30.9 | 68.4 | <5 | [70,71,72,73,74] |
| HHIV-2 | Har. hispanica, Har. vallismortis, Har. japonica | Virulent | Saltern, Italy | Spherical | 80 | Linear dsDNA | 30.6 | 66.5 | - | [2,75] |
| PH1 | Har. hispanica | Persistent | Salt lake, Australia | Spherical | 51 | Linear dsDNA | 28.1 | 67.6 | - | [27] |
| SNJ1 | Natrinema sp. | Temperate | Nnm. sp. (mitomycin C induction) | Spherical | 67 | Circular dsDNA | 16.3 | 48.8-69.7 | <6 | [76,77] |
| His1 | Har. hispanica | Persistent | Salt lake, Australia | Spindle | Head 44 × 77, Tail 7 | Linear dsDNA | 16.5 | 39-40 | <7 | [24,39,78] |
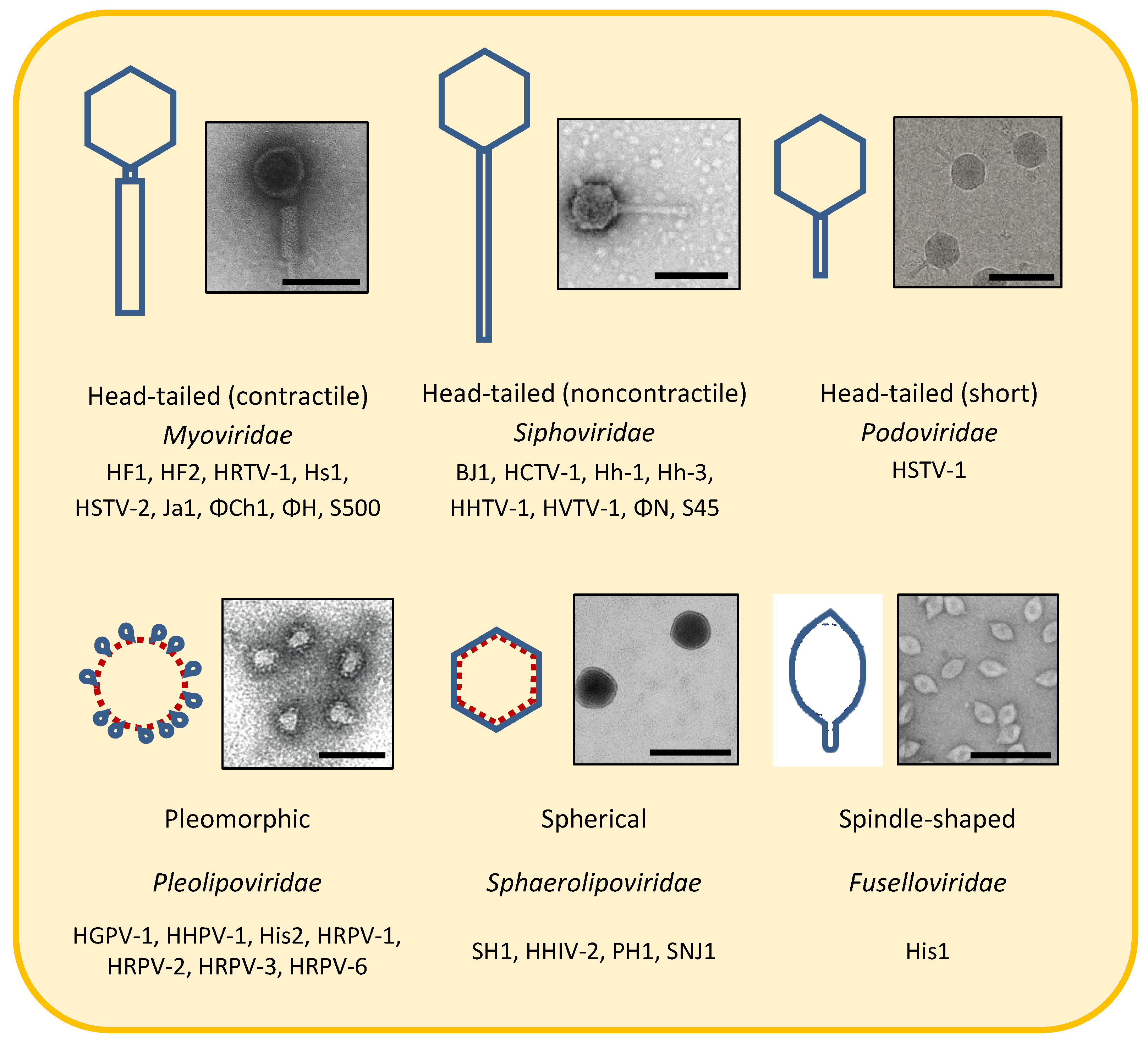
3. Morphotypes
3.1. Head-Tailed Caudoviruses
3.2. Pleolipoviruses
| Subgroups Based on Protein Fragmentation After Virion Dissociation [26] | Subgroups Based on Genome Organization and Replication [68] |
|---|---|
| (1) Soluble fragments only | (1) Use rolling circle replication and contain the replication initiation protein (Rep) |
|
|
| (2) Soluble fragments and one membrane-associated fragment | (2) Use rolling circle replication and do not contain Rep |
|
|
| (3) Membrane-associated fragments only | (3) Use protein-primed replication and have linear genomes |
|
|
3.3. Spherical/Icosahedral Halosphaeroviruses
3.4. Spindle-Shaped/Lemon-Shaped Salterproviruses
4. Macromolecules
4.1. Genomes
4.2. Lipids
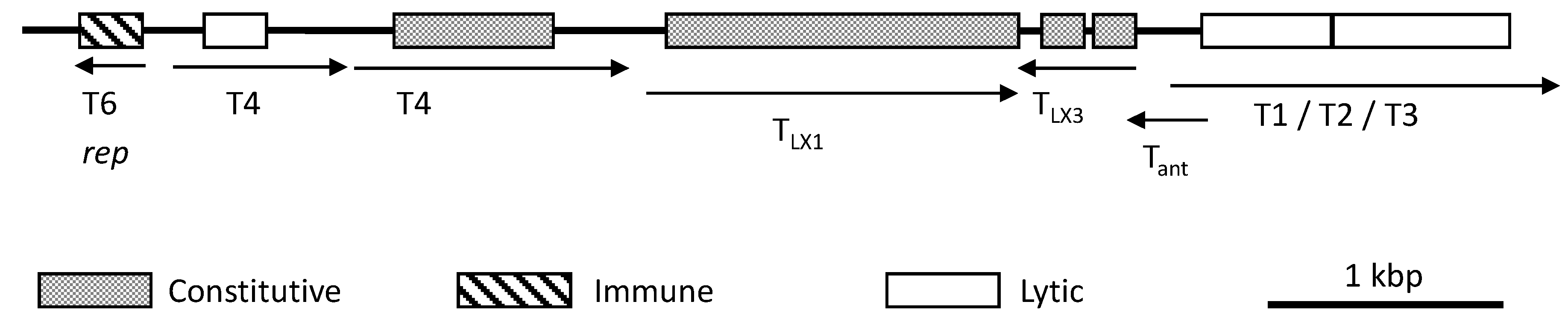
4.3. Structural Proteins of Head-Tailed Haloarchaeoviruses
4.3.1. Capsid Proteins
4.3.2. Tail Proteins
5. Life Cycles
5.1. Adsorption and DNA Ejection
5.2. Replication and Assembly
5.3. Temperate Infection
5.4. Virulent and Persistent Infection
6. Genetic Manipulation and Gene Expression
6.1. Genetic Manipulation
6.2. Gene Regulation
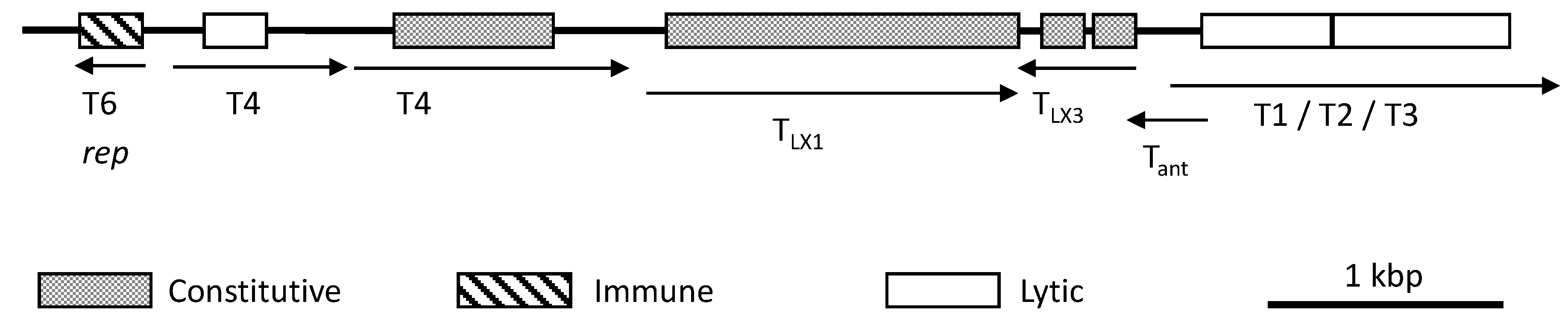
7. Host-Virus Responses
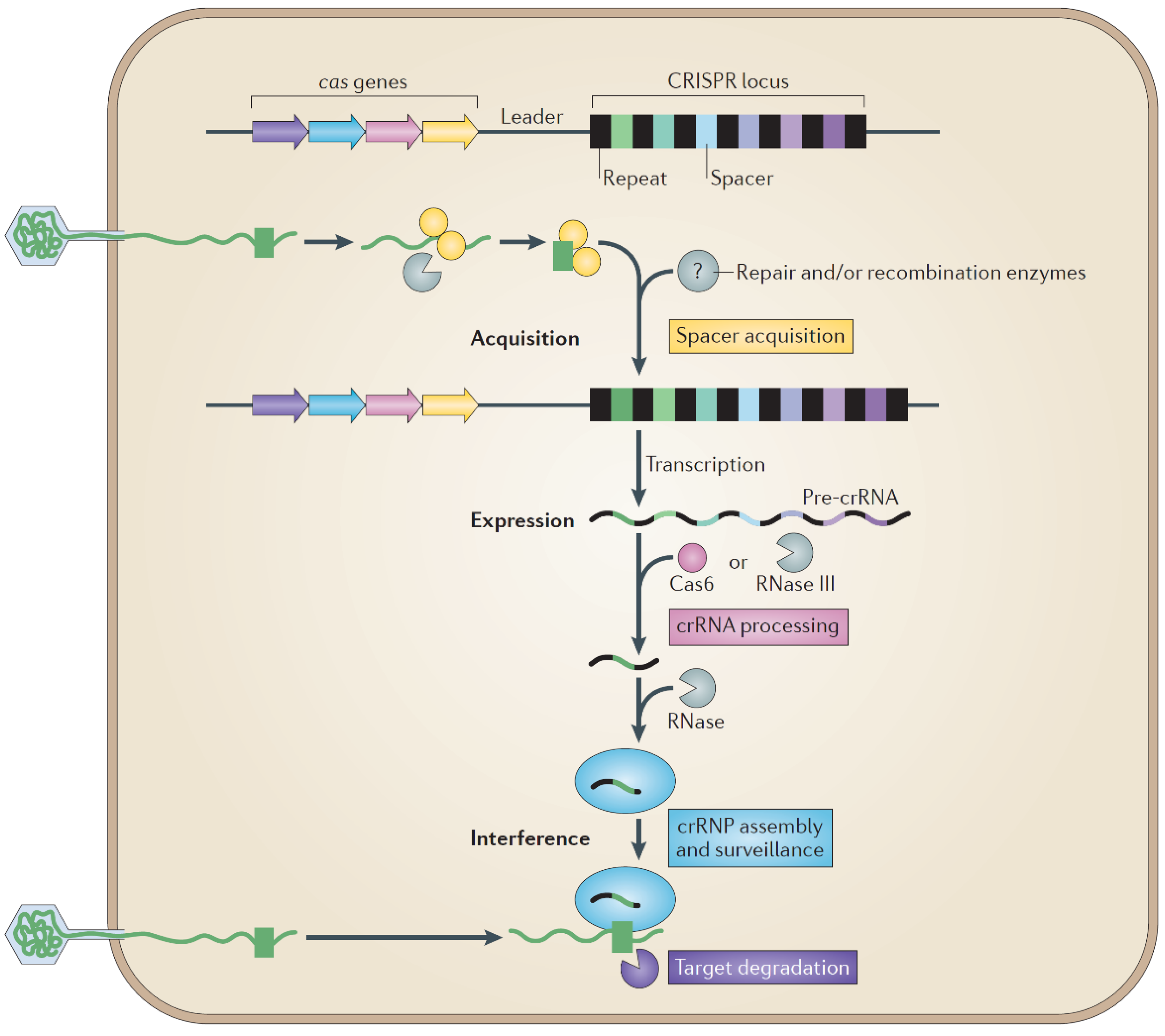
7.1. Host Defense
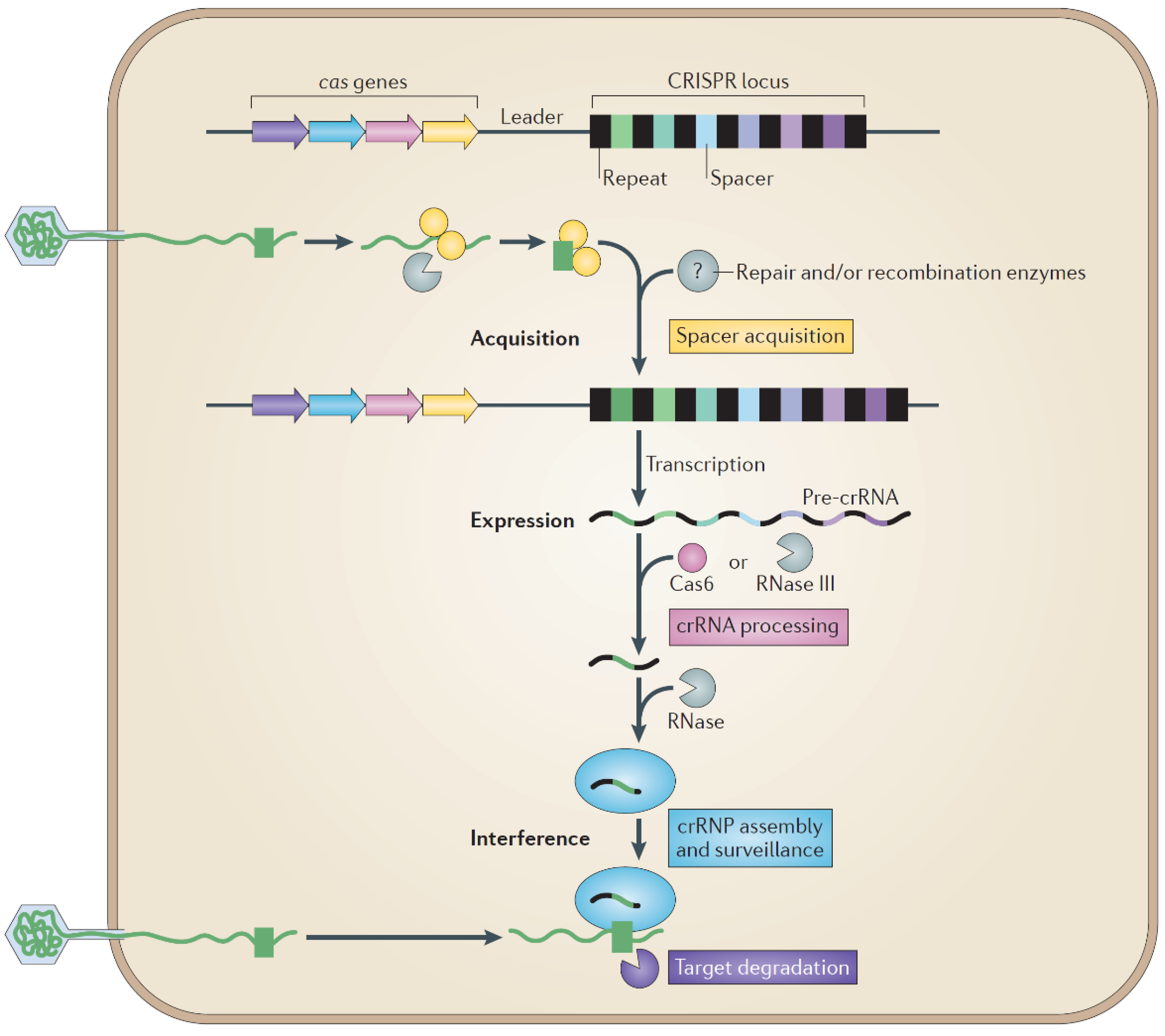
7.2. Host-Range
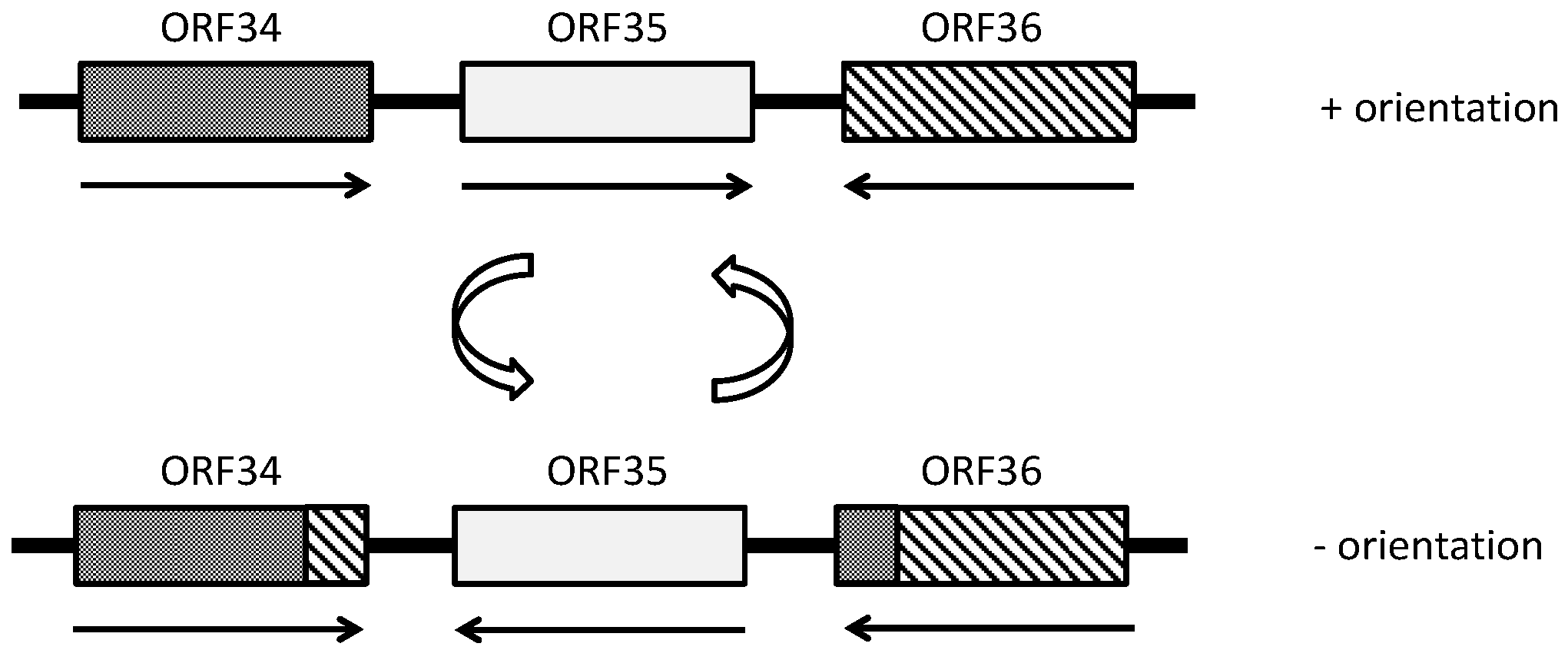
7.3. Haloarchaeovirus Genome Variation
7.4. Salinity, Infection and Evolutionary Strategy
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pina, M.; Bize, A.; Forterre, P.; Prangishvili, D. The archeoviruses. FEMS Microbiol. Rev. 2011, 35, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Roine, E.; Oren, A.; Bamford, D.H.; Oksanen, H.M. Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 2012, 14, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Cavicchioli, R. Archaea-timeline of the third domain. Nat. Rev. Microbiol. 2011, 9, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wais, A.C.; Kon, M.; MacDonald, R.E.; Stollar, B.D. Salt dependent bacteriophage infecting Halobacterium cutirubrum and H. halobium. Nature 1975, 256, 314–315. [Google Scholar] [CrossRef] [PubMed]
- Guixa-Boixareu, N.; Calderón-Paz, J.I.; Heldal, M.; Bratbak, G.; Pedrós-Alió, C. Viral lysis and bacterivory as prokaryotic loss factors along a salinity gradient. Aquat. Microb. Ecol. 1996, 11, 215–227. [Google Scholar] [CrossRef]
- Oren, A. Diversity of halophilic microorganisms: Environments, phylogeny, physiology, and applications. J. Ind. Microbiol. Biotechnol. 2002, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Cho, B.C.; Simpson, A.G. Halocafeteria seosinensis gen. et sp. nov. (Bicosoecida), a halophilic bacterivorous nanoflagellate isolated from a solar saltern. Extremophiles 2006, 10, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, H.; Choi, D.H.; Cho, B.C. Active flagellates grazing on prokaryotes in high salinity waters of a solar saltern. Aquat. Microb. Ecol. 2003, 33, 173–179. [Google Scholar] [CrossRef]
- Casamayor, E.O.; Massana, R.; Benlloch, S.; Ovreas, L.; Diez, B.; Goddard, V.J.; Gasol, J.M.; Joint, I.; Rodriguez-Valera, F.; Pedros-Alio, C. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 2002, 4, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Boujelben, I.; Yarza, P.; Almansa, C.; Villamor, J.; Maalej, S.; Anton, J.; Santos, F. Virioplankton community structure in Tunisian solar salterns. Appl. Environ. Microbiol. 2012, 78, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, I.; Martin-Cuadrado, A.B.; Mojica, F.J.M.; Santos, F.; Mira, A.; Antón, J.; Rodriguez-Valera, F. Reconstructing viral genomes from the environment using fosmid clones: The case of haloviruses. PLoS One 2012, 7, e33802. [Google Scholar] [CrossRef]
- Prangishvili, D.; Forterre, P.; Garrett, R.A. Viruses of the archaea: A unifying view. Nat. Rev. Microbiol. 2006, 4, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, T.; Dundas, I.D. Bacteriophage of Halobacterium salinarium. Nature 1974, 248, 680–681. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Dyall-Smith, M. The Isolation and Study of Viruses of Halophilic Microorganisms. In Extremophiles; Rainey, R.A., Oren, A., Eds.; Elsevier: Waltham, MA, USA, 2006; Volume 35, pp. 681–702. [Google Scholar]
- Dyall-Smith, M.; Tang, S.L.; Bath, C. Haloarchaeal viruses: How diverse are they? Res. Microbiol. 2003, 154, 309–313. [Google Scholar]
- Benlloch, S.; Acinas, S.G.; Anton, J.; Lopez-Lopez, A.; Luz, S.P.; Rodriguez-Valera, F. Archaeal biodiversity in crystallizer ponds from a solar saltern: Culture versus PCR. Microb. Ecol. 2001, 41, 12–19. [Google Scholar] [PubMed]
- Oren, A.; Bratbak, G.; Heldal, M. Occurrence of virus-like particles in the Dead Sea. Extremophiles 1997, 1, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Meyerdierks, A.; Peña, A.; Rosselló-Mora, R.; Amann, R.; Antón, J. Metagenomic approach to the study of halophages: The environmental halophage 1. Environ. Microbiol. 2007, 9, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Bettarel, Y.; Bouvier, T.; Bouvier, C.; Carre, C.; Desnues, A.; Domaizon, I.; Jacquet, S.; Robin, A.; Sime-Ngando, T. Ecological traits of planktonic viruses and prokaryotes along a full-salinity gradient. FEMS Microbiol. Ecol. 2011, 76, 360–372. [Google Scholar] [CrossRef]
- Sime-Ngando, T.; Lucas, S.; Robin, A.; Tucker, K.P.; Colombet, J.; Bettarel, Y.; Desmond, E.; Gribaldo, S.; Forterre, P.; Breitbart, M.; et al. Diversity of virus-host systems in hypersaline Lake Retba, Senegal. Environ. Microbiol. 2011, 13, 1956–1972. [Google Scholar] [CrossRef]
- Prangishvili, D.; Garrett, R.A. Viruses of hyperthermophilic Crenarchaea. Trends Microbiol. 2005, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 2006, 350, 228–239. [Google Scholar]
- Tang, S.L.; Nuttall, S.; Dyall-Smith, M. Haloviruses HF1 and HF2: Evidence for a recent and large recombination event. J. Bacteriol. 2004, 186, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Atanasova, N.S.; Manole, V.; Liljeroos, L.; Butcher, S.J.; Oksanen, H.M.; Bamforda, D.H. Virion architecture unifies globally distributed pleolipoviruses infecting halophilic Archaea. J. Virol. 2012, 86, 5067–5078. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Tang, S.L.; Chen, C.P.; Chiang, P.W.; Hong, M.J.; Dyall-Smith, M. Ph1: An archaeovirus of Haloarcula hispanica related to SH1 and HHIV-2. Archaea 2013, 2013, 456318. [Google Scholar] [CrossRef]
- Bamford, D.H. Do viruses form lineages across different domains of life? Res. Microbiol. 2003, 154, 231–236. [Google Scholar]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure unifies the viral universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef]
- Krupovič, M.; Bamford, D.H. Virus evolution: How far does the double β-barrel viral lineage extend? Nat. Rev. Microbiol. 2008, 6, 941–948. [Google Scholar]
- Jalasvuori, M; Bamford, J.K.H. Viruses and life: Can there be one without the other? J. Cosmol. 2010, 10, 3446–3454. [Google Scholar]
- Bamford, D.H.; Grimes, J.M.; Stuart, D.I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of highly mosaic mycobacteriophage genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef]
- Papke, R.T.; Doolittle, W.F. Phage evolution: New worlds of genomic diversity. Curr. Biol. 2003, 13, R606–R607. [Google Scholar] [CrossRef] [PubMed]
- Roine, E.; Kukkaro, P.; Paulin, L.; Laurinavičius, S.; Domanska, A.; Somerharju, P.; Bamford, D.H. New, closely related haloarchaeal viral elements with different nucleic acid types. J. Virol. 2010, 84, 3682–3689. [Google Scholar] [CrossRef] [PubMed]
- Krupovič, M.; Bamford, D.H. Order to the viral universe. J. Virol. 2010, 84, 12476–12479. [Google Scholar] [CrossRef]
- Benson, S.D.; Bamford, J.K.H.; Bamford, D.H.; Burnett, R.M. Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell 2004, 16, 673–685. [Google Scholar]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Modified coat protein forms the flexible spindle-shaped virion of haloarchaeal virus His1. Environ. Microbiol. 2013, 15, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Jaatinen, S.T.; Happonen, L.J.; Laurinmaki, P.; Butcher, S.J.; Bamford, D.H. Biochemical and structural characterisation of membrane-containing icosahedral dsDNA bacteriophages infecting thermophilic Thermus thermophilus. Virology 2008, 379, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Jalasvuori, M.; Jaatinen, S.T.; Laurinavicius, S.; Ahola-Iivarinen, E.; Kalkkinen, N.; Bamford, D.H.; Bamford, J.K. The closest relatives of icosahedral viruses of thermophilic Bacteria are among viruses and plasmids of the halophilic Archaea. J. Virol. 2009, 83, 9388–9397. [Google Scholar] [CrossRef] [PubMed]
- Jalasvuori, M.; Pawlowski, A.; Bamford, J.K. A unique group of virus-related, genome-integrating elements found solely in the bacterial family Thermaceae and the archaeal family Halobacteriaceae. J. Bacteriol. 2010, 192, 3231–3234. [Google Scholar] [CrossRef]
- Jäälinoja, H.T.; Roine, E.; Laurinmäki, P.; Kivelä, H.M.; Bamford, D.H.; Butcher, S.J. Structure and host-cell interaction of SH1, a membrane-containing, halophilic euryarchaeal virus. Proc. Natl. Acad. Sci. USA 2008, 105, 8008–8013. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M. Halobacterial viruses (haloviruses) summary table. Available online: http://www.haloarchaea.com/research/halovirus/VirusTable.html (accessed on 28 May 2013).
- Nuttall, S.D.; Dyall-Smith, M.L. HF1 and HF2: Novel bacteriophages of halophilic Archaea. Virology 1993, 197, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.D.; Dyall-Smith, M.L. Halophage HF2: Genome organization and replication strategy. J. Virol. 1995, 69, 2322–2327. [Google Scholar] [PubMed]
- Senčilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Os̈terlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Kukkaro, P.; Bamford, D.H. Virus-host interactions in environments with a wide range of ionic strengths. Environ. Microbiol. Rep. 2009, 1, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, T.; Dundas, I.D. Persisting phage infection in Halobacterium salinarium str. 1. J. Gen. Virol. 1980, 47, 29–36. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Butcher, S.J.; Bamford, D.H.; Hendrix, R.W. Insights into head-tailed viruses infecting extremely halophilic Archaea. J. Virol. 2013, 87, 3248–3260. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Baranyi, U.; Klein, R.; Sulzner, M.; Luo, C.; Wanner, G.; Krüger, D.H.; Lubitz, W. Characterization of Natronobacterium magadii phage φCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 1997, 23, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Baranyi, U.; Rössler, N.; Greineder, B.; Scholz, H.; Witte, A. Natrialba magadii virus φCh1: First complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol. Microbiol. 2002, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
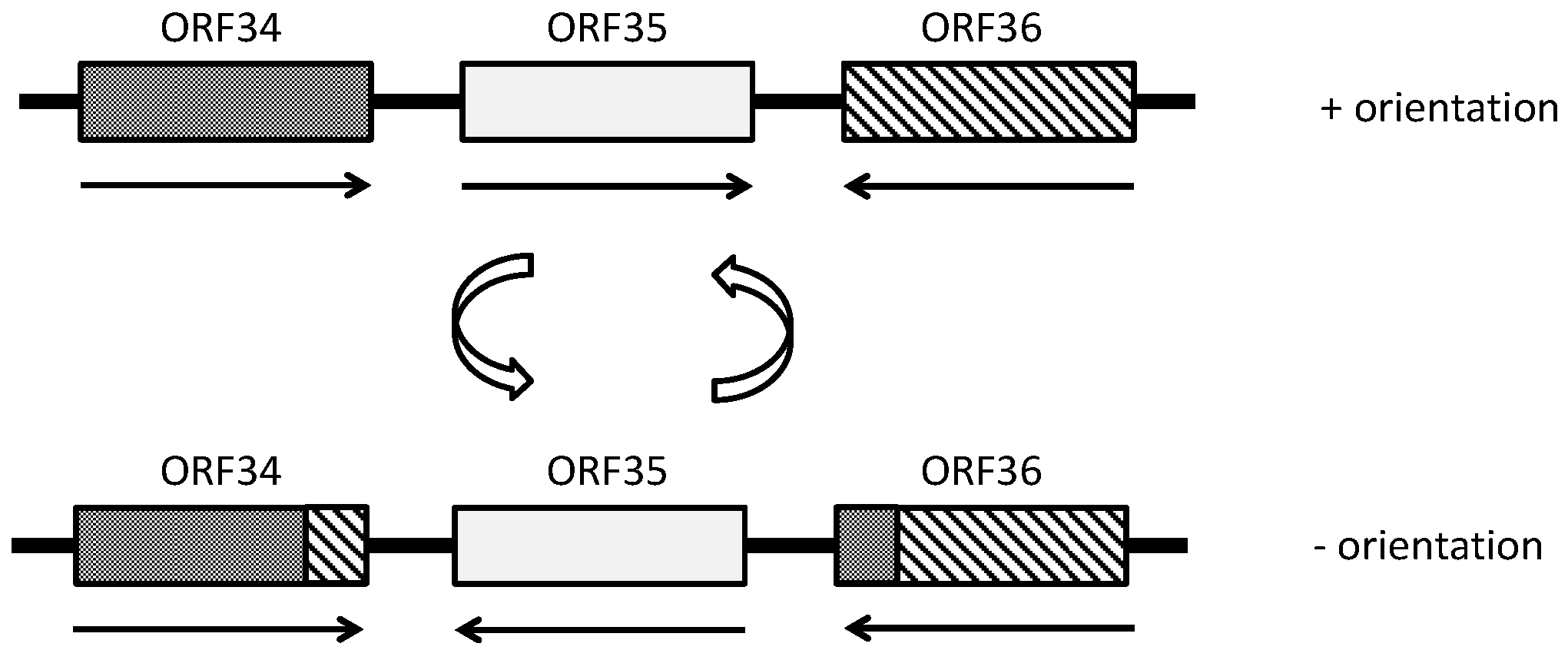
- Rössler, N.; Klein, R.; Scholz, H.; Witte, A. Inversion within the haloalkaliphilic virus φCh1 DNA results in differential expression of structural proteins. Mol. Microbiol. 2004, 52, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H.; Zillig, W.; Pfäffle, M.; Schnabel, R.; Michel, H.; Delius, H. Halobacterium halobium phage øH. EMBO J. 1982, 1, 87–92. [Google Scholar] [PubMed]
- Schnabel, H.; Schramm, E.; Schnabel, R.; Zillig, W. Structural variability in the genome of phage φH of Halobacterium halobium. Mol. Gen. Genet. 1982, 188, 370–377. [Google Scholar] [CrossRef]
- Gropp, F.; Palm, P.; Zillig, W. Expression and regulation of Halobacterium halobium phage φH genes. Can. J. Microbiol. 1989, 35, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo studies on the effects of immunity genes on early lytic transcription in the Halobacterium salinarium phage φH. Mol. Gen. Genet. 1992, 235, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Antisense RNA mediates transcriptional processing in an archaebacterium, indicating a novel kind of RNase activity. Mol. Microbiol. 1993, 7, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo and in vitro analysis of transcription of the l region from the Halobacterium salinarium phage øH: Definition of a repressor-enhancing gene. Virology 1993, 195, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Gene regulation in halophage φH; more than promoters. Syst. Appl. Microbiol. 1994, 16, 591–596. [Google Scholar] [CrossRef]
- Stolt, P.; Zillig, W. Transcription of the halophage φH repressor gene is abolished by transcription from an inversely oriented lytic promoter. FEBS Lett. 1994, 344, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.L.; Wais, A.C. Ecophysiology of bacteriophage S5100 infecting Halobacterium cutirubrum. Appl. Environ. Microbiol. 1990, 56, 3605–3608. [Google Scholar] [PubMed]
- Pagaling, E.; Haigh, R.D.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Sequence analysis of an archaeal virus isolated from a hypersaline lake in inner Mongolia, China. BMC Genomics 2007, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Pauling, C. Bacteriophages of Halobacterium halobium: Isolation from fermented fish sauce and primary characterization. Can. J. Microbiol. 1982, 28, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F.; Cheney, R.; Pauling, C. Bacteriophages of Halobacterium halobium: Virion DNAs and proteins. Can. J. Microbiol. 1983, 29, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang-Wenke, H.; Oesterhelt, D. Isolation of a halobacterial phage with a fully cytosine-methylated genome. Mol. Gen. Genet. 1988, 211, 407–414. [Google Scholar] [CrossRef]
- Daniels, L.L.; Wais, A.C. Restriction and modification of halophage S45 in Halobacterium. Curr. Micriobiol. 1984, 10, 133–136. [Google Scholar] [CrossRef]
- Senčilo, A.; Paulin, L.; Kellner, S.; Helm, M.; Roine, E. Related haloarchaeal pleomorphic viruses contain different genome types. Nucleic Acids Res. 2012, 40, 5523–5534. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Roine, E.; Paulin, L.; Kalkkinen, N.; Bamford, D.H. An ssDNA virus infecting Archaea: A new lineage of viruses with a membrane envelope. Mol. Microbiol. 2009, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Dyall-Smith, M.L. Transfection of haloarchaea by the DNAs of spindle and round haloviruses and the use of transposon mutagenesis to identify non-essential regions. Mol. Microbiol. 2008, 70, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.H.; Ravantti, J.J.; Ronnholm, G.; Laurinavicius, S.; Kukkaro, P.; Dyall-Smith, M.; Somerharju, P.; Kalkkinen, N.; Bamford, J.K. Constituents of SH1, a novel lipid-containing virus infecting the halophilic euryarchaeon Haloarcula hispanica. J. Virol. 2005, 79, 9097–9107. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Kukkaro, P.; Bamford, J.K.H.; Bath, C.; Kivelä, H.M.; Dyall-Smith, M.L.; Bamford, D.H. SH1: A novel, spherical halovirus isolated from an Australian hypersaline lake. Virology 2005, 335, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, H.M.; Roine, E.; Kukkaro, P.; Laurinavičius, S.; Somerharju, P.; Bamford, D.H. Quantitative dissociation of archaeal virus SH1 reveals distinct capsid proteins and a lipid core. Virology 2006, 356, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Russ, B.E.; Yang, J.; Dyall-Smith, M.L. The transcription programme of the protein-primed halovirus SH1. Microbiology 2008, 154, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, S.T.; Penttinen, R.K.; Vilén, S.T.; Jalasvuori, M.; Rönnholm, G.; Bamford, J.K.H.; Bamford, D.H.; Oksanen, H.M. Closely related archaeal Haloarcula hispanica icosahedral viruses HHIV-2 and SH1 have nonhomologous genes encoding host recognition functions. J. Virol. 2012, 86, 4734–4742. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Chen, J.; Sun, D.; Chen, D.; Yang, Y.; Shen, P.; Chen, X. Induction and preliminary characterization of a novel halophage SNJ1 from lysogenic Natrinema sp. F5. Can. J. Microbiol. 2007, 53, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, Y.; Wang, S.; Yang, D.; Cheng, Y.; Hu, J.; Chen, J.; Mei, Y.; Shen, P.; Bamford, D.H.; et al. Temperate membrane-containing halophilic archaeal virus SNJ1 has a circular dsDNA genome identical to that of plasmid pHH205. Virology 2012, 434, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bath, C.; Dyall-smith, M.L. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula hispanica. J. Virol. 1998, 72, 9392–9395. [Google Scholar] [PubMed]
- Ventosa, A.; Oren, A. Halobacterium salinarum nom. corrig, a name to replace Halobacterium salinarium (elazari-volcani) and to include Halobacterium halobium and Halobacterium cutirubrum. Int. J. Syst. Bacteriol. 1996, 46, 347. [Google Scholar] [CrossRef]
- Euzeby, J.P. List of prokaryotic names with standing in nomenclature. Available online: http://www.bacterio.net/-abbreviations.html#halobacteriaceae (accessed on 5 September 2014).
- Ackermann, H.W. Tailed bacteriophages: The order Caudovirales. Adv. Virus. Res. 1998, 51, 135–201. [Google Scholar] [PubMed]
- Hendrix, R.W. Bacteriophage HK97: Assembly of the capsid and evolutionary connections. Adv. Virus. Res. 2005, 64, 1–14. [Google Scholar] [PubMed]
- Chang, J.R.; Spilman, M.S.; Rodenburg, C.M.; Dokland, T. Functional domains of the bacteriophage P2 scaffolding protein: Identification of residues involved in assembly and protease activity. Virology 2009, 384, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Krupovič, M.; Forterre, P.; Bamford, D.H. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of Bacteria. J. Mol. Biol. 2010, 397, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Demina, T.A.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Archaeal viruses and bacteriophages: Comparisons and contrasts. Trends Microbiol. 2014, 22, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D. The wonderful world of archaeal viruses. Annu. Rev. Microbiol. 2013, 67, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Spang, A.; Gribaldo, S.; Forterre, P.; Schleper, C. A thaumarchaeal provirus testifies for an ancient association of tailed viruses with archaea. Biochem. Soc. Trans. 2011, 39, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Senčilo, A.; Roine, E. A glimpse of the genomic diversity of haloarchaeal tailed viruses. Front. Microbiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Laurinavičius, S.; Sund, J.; Roine, E.; Bamford, D.H. The single-stranded DNA genome of novel archaeal virus Halorubrum pleomorphic virus 1 is enclosed in the envelope decorated with glycoprotein spikes. J. Virol. 2010, 84, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Dybvig, K.; Nowak, J.A.; Sladek, T.L.; Maniloff, J. Identification of an enveloped phage, mycoplasma virus L172, that contains a 14-kilobase single-stranded DNA genome. J. Virol. 1985, 53, 384–390. [Google Scholar] [PubMed]
- Pawlowski, A.; Rissanen, I.; Bamford, J.K.; Krupovic, M.; Jalasvuori, M. Gammasphaerolipovirus, a newly proposed bacteriophage genus, unifies viruses of halophilic Archaea and thermophilic Bacteria within the novel family Sphaerolipoviridae. Arch. Virol. 2014, 159, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Quemin, E.R.; Bamford, D.H.; Forterre, P.; Prangishvili, D. Unification of the globally distributed spindle-shaped viruses of the Archaea. J. Virol. 2014, 88, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Haring, M.; Vestergaard, G.; Rachel, R.; Chen, L.; Garrett, R.A.; Prangishvili, D. Virology: Independent virus development outside a host. Nature 2005, 436, 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Scheele, U.; Erdmann, S.; Ungewickell, E.J.; Felisberto-Rodrigues, C.; Ortiz-Lombardia, M.; Garrett, R.A. Chaperone role for proteins P618 and P892 in the extracellular tail development of Acidianus two-tailed virus. J. Virol. 2011, 85, 4812–4821. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Chen, L.; Huang, X.; Luo, Y.; She, Q.; Huang, L. Sulfolobus tengchongensis spindle-shaped virus STSV1: Virus-host interactions and genomic features. J. Virol. 2005, 79, 8677–8686. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, S.; Chen, B.; Huang, X.; Deng, L.; Liu, C.; Shah, S.A.; Le Moine Bauer, S.; Sobrino, C.L.; Wang, H.; Wei, Y.; et al. A novel single-tailed fusiform Sulfolobus virus STSV2 infecting model Sulfolobus species. Extremophiles 2014, 18, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Allen, E.E.; Heidelberg, K.B.; Banfielda, J.F. Dynamic viral populations in hypersaline systems as revealed by metagenomic assembly. Appl. Environ. Microbiol. 2012, 78, 6309–6320. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Andrade, K.; Thomas, B.C.; Norman, A.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Virus-host and CRISPR dynamics in Archaea-dominated hypersaline Lake Tyrrell, Victoria, Australia. Archaea 2013, 2013, 370871. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.L.; Nuttall, S.; Ngui, K.; Fisher, C.; Lopez, P.; Dyall-Smith, M. HF2: A double-stranded DNA tailed haloarchaeal virus with a mosaic genome. Mol. Microbiol. 2002, 44, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Rössler, N.; Iro, M.; Scholz, H.; Witte, A. Haloarchaeal myovirus φCh1 harbours a phase variation system for the production of protein variants with distinct cell surface adhesion specificities. Mol. Microbiol. 2012, 83, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Yarza, P.; Parro, V.; Briones, C.; Anton, J. The metavirome of a hypersaline environment. Environ. Microbiol. 2010, 12, 2965–2976. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; Ugalde, J.A.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012, 6, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Pasic, L.; Fernandez, A.B.; Martin-Cuadrado, A.B.; Mizuno, C.M.; McMahon, K.D.; Papke, R.T.; Stepanauskas, R.; Rodriguez-Brito, B.; Rohwer, F.; et al. New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Stepanauskas, R.; Mohan, N.R.; Papke, R.T. Cell sorting analysis of geographically separated hypersaline environments. Extremophiles 2013, 17, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Cavicchioli, R. Extremophiles and the search for extraterrestrial life. Astrobiology 2002, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
- DasSarma, S. Extreme halophiles are models for astrobiology. Microbe 2006, 1, 120–126. [Google Scholar]
- Pasic, L.; Rodriguez-Mueller, B.; Martin-Cuadrado, A.B.; Mira, A.; Rohwer, F.; Rodriguez-Valera, F. Metagenomic islands of hyperhalophiles: The case of Salinibacter ruber. BMC Genomics 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Roine, E.; Bamford, D.H. Lipids of archaeal viruses. Archaea 2012, 2012, 384919. [Google Scholar] [CrossRef] [PubMed]
- Katsura, I.; Hendrix, R.W. Length determination in bacteriophage lambda tails. Cell 1984, 39, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Wang, I.N. Bacteriophage adsorption rate and optimal lysis time. Genetics 2008, 180, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Russ, B.E.; Dyall-Smith, M.L. Virus-host interactions in salt lakes. Curr. Opin. Microbiol. 2007, 10, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Hanhijärvi, K.J.; Ziedaite, G.; Pietilä, M.K.; Hæggström, E.; Bamford, D.H. DNA ejection from an archaeal virus - a single-molecule approach. Biophys. J. 2013, 104, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Grayson, P.; Han, L.; Winther, T.; Phillips, R. Real-time observations of single bacteriophage λ DNA ejections in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 14652–14657. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H. An immune strain of Halobacterium halobium carries the invertible L segment of phage φH as a plasmid. Proc. Natl. Acad. Sci. USA 1984, 81, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Lobocka, M.B.; Rose, D.J.; Plunkett, G., III; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H.; Zillig, W. Circular structure of the genome of phage φH in a lysogenic Halobacterium halobium. Mol. Gen. Genet. 1984, 193, 422–426. [Google Scholar] [CrossRef]
- DeMaere, M.Z.; Williams, T.J.; Allen, M.A.; Brown, M.V.; Gibson, J.A.; Rich, J.; Lauro, F.M.; Dyall-Smith, M.; Davenport, K.W.; Woyke, T.; et al. High level of intergenera gene exchange shapes the evolution of haloarchaea in an isolated Antarctic lake. Proc. Natl. Acad. Sci. USA 2013, 110, 16939–16944. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, C.; Zhao, Z.; Yang, Z.L. Genome sequence of Halorubrum sp. strain T3, an extremely halophilic archaeon harboring a virus-like element. J. Bacteriol. 2012, 194, 6608–6609. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, C.; Xu, J.P.; Yang, Z.L. Molecular characterization of PHRDV1, a new virus-like mobile genetic element closely related to pleomorphic viruses in haloarchaea. Extremophiles 2014, 18, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.W.; Doolittle, W.F. Efficient transfection of the archaebacterium Halobacterium halobium. J. Bacteriol. 1987, 169, 1341–1344. [Google Scholar] [PubMed]
- Cline, S.W.; Lam, W.L.; Charlebois, R.L.; Schalkwyk, L.C.; Doolittle, W.F. Transformation methods for halophilic archaebacteria. Can. J. Microbiol. 1989, 35, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Blaseio, U.; Pfeifer, F. Transformation of Halobacterium halobium: Development of vectors and investigation of gas vesicle synthesis. Proc. Natl. Acad. Sci. USA 1990, 87, 6772–6776. [Google Scholar] [CrossRef] [PubMed]
- Iro, M.; Klein, R.; Gálos, B.; Baranyi, U.; Rössler, N.; Witte, A. The lysogenic region of virus φCh1: Identification of a repressor-operator system and determination of its activity in halophilic Archaea. Extremophiles 2007, 11, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Mevarech, M. Archaeal genetics—the third way. Nat. Rev. Genet. 2005, 6, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Cuadros-Orellana, S.; Martin-Cuadrado, A.B.; Legault, B.; D’Auria, G.; Zhaxybayeva, O.; Papke, R.T.; Rodriguez-Valera, F. Genomic plasticity in prokaryotes: The case of the square haloarchaeon. ISME J. 2007, 1, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Pyatibratov, M.G.; Beznosov, S.N.; Rachel, R.; Tiktopulo, E.I.; Surin, A.K.; Syutkin, A.S.; Fedorov, O.V. Alternative flagellar filament types in the haloarchaeon Haloarcula marismortui. Can. J. Microbiol. 2008, 54, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef]
- Rousseau, C.; Gonnet, M.; Le Romancer, M.; Nicolas, J. CRISPI: A CRIPSR interactive database. Bioinformatics 2009, 25, 3317–3318. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Wolf, Y.I. Is evolution Darwinian or/and Lamarckian? Biol. Direct 2009, 4, 42. [Google Scholar] [CrossRef]
- Maier, L.K.; Fischer, S.; Stoll, B.; Brendel, J.; Pfeiffer, F.; Dyall-Smith, M.; Marchfelder, A. The immune system of halophilic Archaea. Mob. Genet. Elements 2012, 2, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Van der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in Bacteria and Archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, S.; Westra, E.R.; van der Oost, J.; Brouns, S.J. Clustered regularly interspaced short palindromic repeats (CRISPRs): The hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol. Chem. 2011, 392, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.A.; Vestergaard, G.; Shah, S.A. Archaeal CRISPR-based immune systems: Exchangeable functional modules. Trends Microbiol. 2011, 19, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, G.; Garrett, R.A.; Shah, S.A. CRISPR adaptive immune systems of Archaea. RNA Biol. 2014, 11, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Maier, L.K.; Stoll, B.; Brendel, J.; Fischer, E.; Pfeiffer, F.; Dyall-Smith, M.; Marchfelder, A. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J. Biol. Chem. 2012, 287, 33351–33365. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.K.; Stoll, B.; Brendel, J.; Fischer, S.; Pfeiffer, F.; Dyall-Smith, M.; Marchfelder, A. The ring of confidence: A haloarchaeal CRISPR/Cas system. Biochem. Soc. Trans. 2013, 41, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, H.; Han, J.; Liu, J.; Wang, R.; Zhao, D.; Zhou, J.; Xiang, H. Characterization of CRISPR RNA biogenesis and Cas6 cleavage-mediated inhibition of a provirus in the haloarchaeon Haloferax mediterranei. J. Bacteriol. 2013, 195, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Fullmer, M.S.; Soucy, S.M.; Swithers, K.S.; Makkay, A.M.; Wheeler, R.; Ventosa, A.; Gogarten, J.P.; Papke, R.T. Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonte, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Naor, A.; Lapierre, P.; Mevarech, M.; Papke, R.T.; Gophna, U. Low species barriers in halophilic Archaea and the formation of recombinant hybrids. Curr. Biol. 2012, 22, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Moreno-Paz, M.; Meseguer, I.; López, C.; Rosselló-Mora, R.; Parro, V.; Antón, J. Metatranscriptomic analysis of extremely halophilic viral communities. ISME J. 2011, 5, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.L.; Pfeiffer, F.; Klee, K.; Palm, P.; Gross, K.; Schuster, S.C.; Rampp, M.; Oesterhelt, D. Haloquadratum walsbyi: Limited diversity in a global pond. PLoS One 2011, 6, e20968. [Google Scholar] [CrossRef] [PubMed]
- Wais, A.C.; Daniels, L.L. Populations of bacteriophage infecting Halobacterium in a transient brine pool. FEMS Microbiol. Ecol. 1985, 31, 323–326. [Google Scholar] [CrossRef]
- Bertani, G. Transduction-like gene transfer in the methanogen Methanococcus voltae. J. Bacteriol. 1999, 181, 2992–3002. [Google Scholar] [PubMed]
- Lang, A.S.; Zhaxybayeva, O.; Beatty, J.T. Gene transfer agents: Phage-like elements of genetic exchange. Nat. Rev. Microbiol. 2012, 10, 472–482. [Google Scholar] [PubMed]
- Williams, D.; Gogarten, J.P.; Papke, R.T. Quantifying homologous replacement of loci between haloarchaeal species. Genome Biol. Evol. 2012, 4, 1223–1244. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luk, A.W.S.; Williams, T.J.; Erdmann, S.; Papke, R.T.; Cavicchioli, R. Viruses of Haloarchaea. Life 2014, 4, 681-715. https://doi.org/10.3390/life4040681
Luk AWS, Williams TJ, Erdmann S, Papke RT, Cavicchioli R. Viruses of Haloarchaea. Life. 2014; 4(4):681-715. https://doi.org/10.3390/life4040681
Chicago/Turabian StyleLuk, Alison W. S., Timothy J. Williams, Susanne Erdmann, R. Thane Papke, and Ricardo Cavicchioli. 2014. "Viruses of Haloarchaea" Life 4, no. 4: 681-715. https://doi.org/10.3390/life4040681
APA StyleLuk, A. W. S., Williams, T. J., Erdmann, S., Papke, R. T., & Cavicchioli, R. (2014). Viruses of Haloarchaea. Life, 4(4), 681-715. https://doi.org/10.3390/life4040681