
Epigenetic Profiling of Cell-Free DNA in Cerebrospinal Fluid: A Novel Biomarker Approach for Metabolic Brain Diseases

 , , , , and
, , , , and
Abstract
1. Introduction
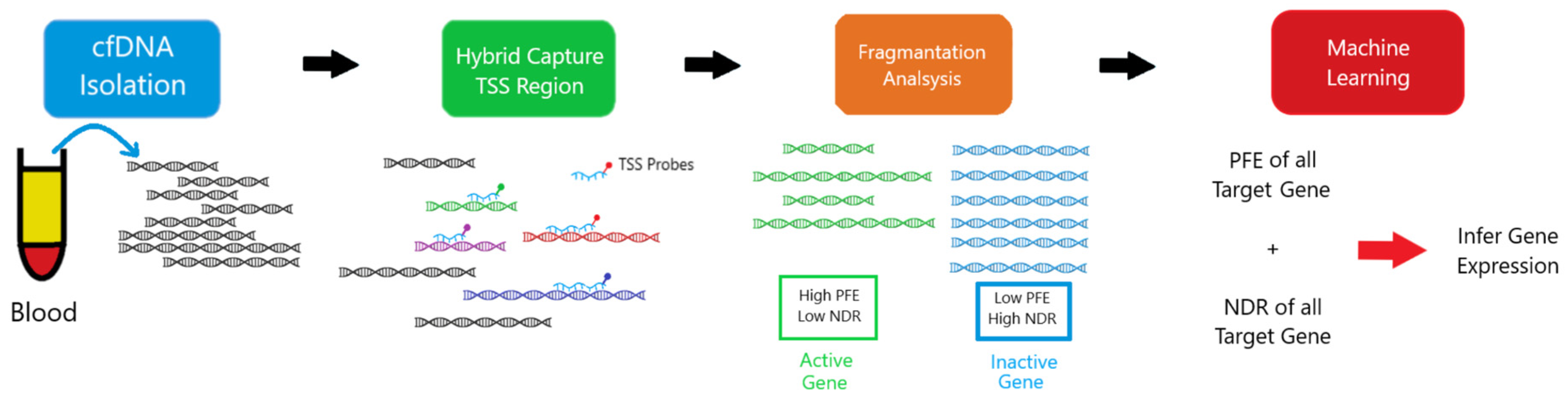
2. Methodology
3. Metabolic Brain Diseases: Clinical Spectrum and Diagnostic Imperatives
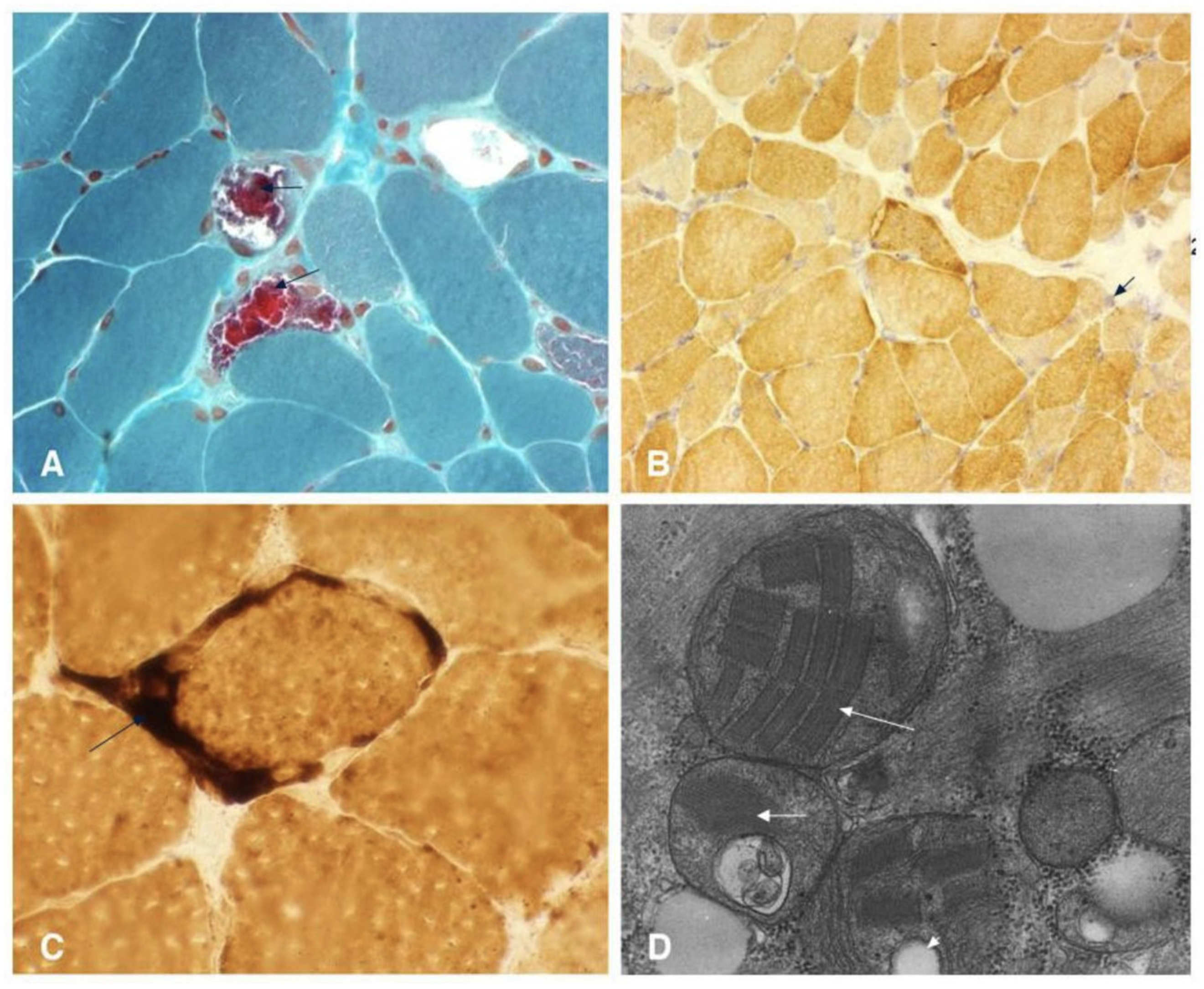
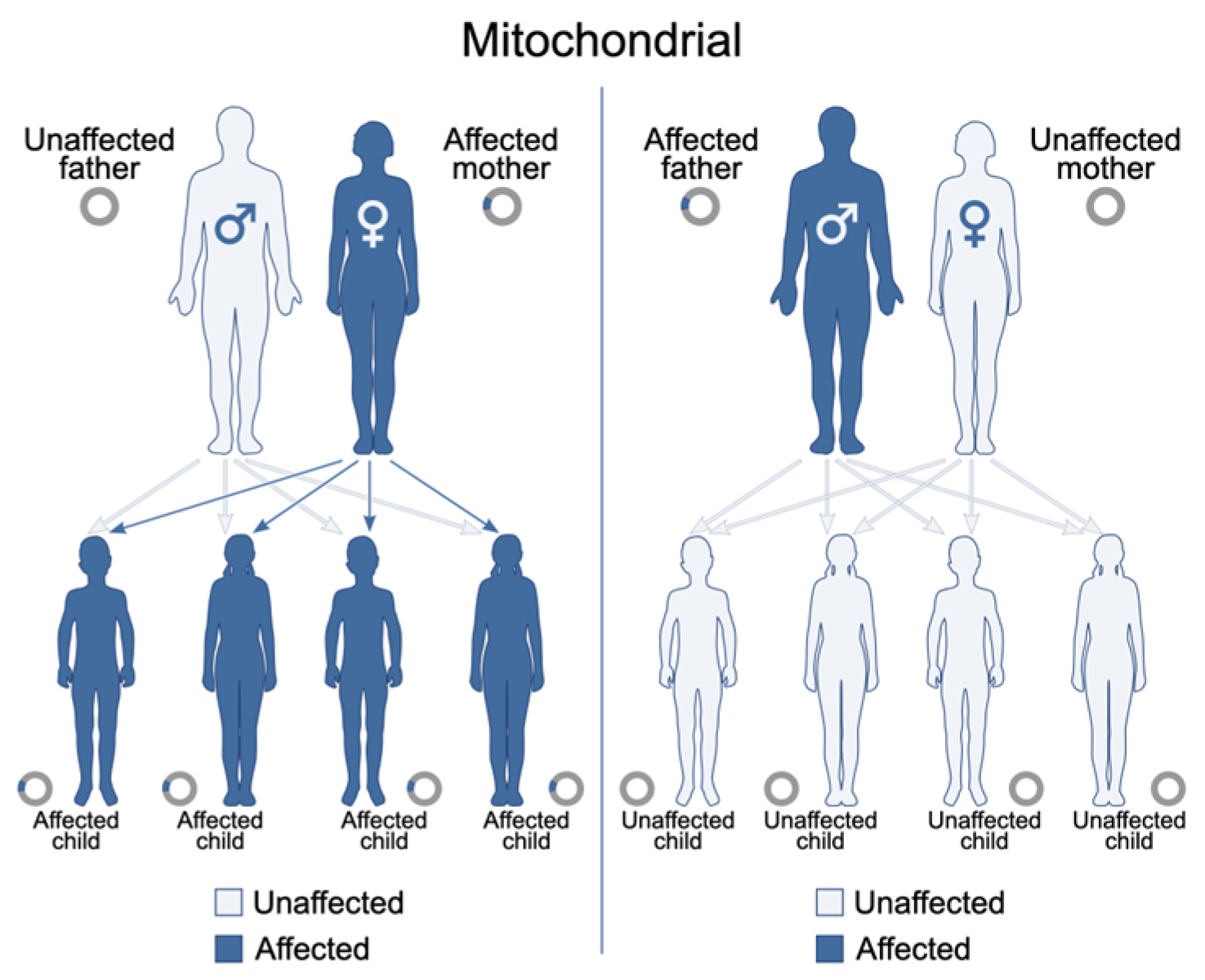
3.1. Mitochondrial Encephalopathies (e.g., MELAS)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Category | Example Diseases | Key Clinical Manifestations | Common Diagnostic Methods | Key Diagnostic Limitations |
|---|---|---|---|---|
| Mitochondrial Encephalopathies [21,24,25,27,28] | MELAS | Stroke-like episodes (pre-40), encephalopathy (seizures/dementia), lactic acidosis (blood/CSF), myopathy, recurrent headaches, short stature, hearing impairment. | Increased Blood/CSF lactate, Increased lactate: pyruvate, muscle biopsy (ragged red fibers), mtDNA genetic testing. | Non-specific lactic acidosis, heteroplasmy/tissue distribution challenges for genetic testing, muscle biopsy not always definitive, neuroimaging non-specific. |
| Lysosomal Storage Diseases [29,30,31,32,33] | Niemann-Pick (Types A, B, C), Gaucher (Types 1, 2, 3) | Early onset: Hepatosplenomegaly, developmental delay, hypotonia, jaundice. Later onset: Progressive neurodegeneration (ataxia, dementia, seizures), vertical gaze palsy (NPC), organomegaly, musculoskeletal issues, psychiatric symptoms (NPC). | Biochemical enzyme assays, mutational analysis (e.g., SMPD1, NPC1, NPC2, GBA1 genes), oxysterols (NPC). | High index of suspicion needed, biomarker non-specificity (oxysterols), sample stability issues, genetic testing complexities (polymorphism, VUS), non-specific neuroimaging, clinical assessment difficulties. |
| Glucose Metabolism Disorders [34,35,36,37,38] | GLUT1 Deficiency Syndrome, Diabetes-Associated Cognitive Decline | GLUT1DS: Early-onset seizures, developmental delay, cognitive impairment, movement disorders, microcephaly, speech/language issues. DCD: Attention/memory/executive function deficits, visuospatial decline, increased dementia risk. | GLUT1DS: CSF glucose/lactate, SLC2A1 genetic testing. DCD: Neurocognitive assessment, blood glucose/HbA1c. | GLUT1DS: Symptom variability/underdiagnosis, CSF glucose/lactate can be near normal, genetic mimics (PURA, HK1), functional assay availability. DCD: Non-specific cognitive symptoms, early metabolic changes precede symptoms, overlap/mimicry with AD/other dementias, complex pathophysiology. |
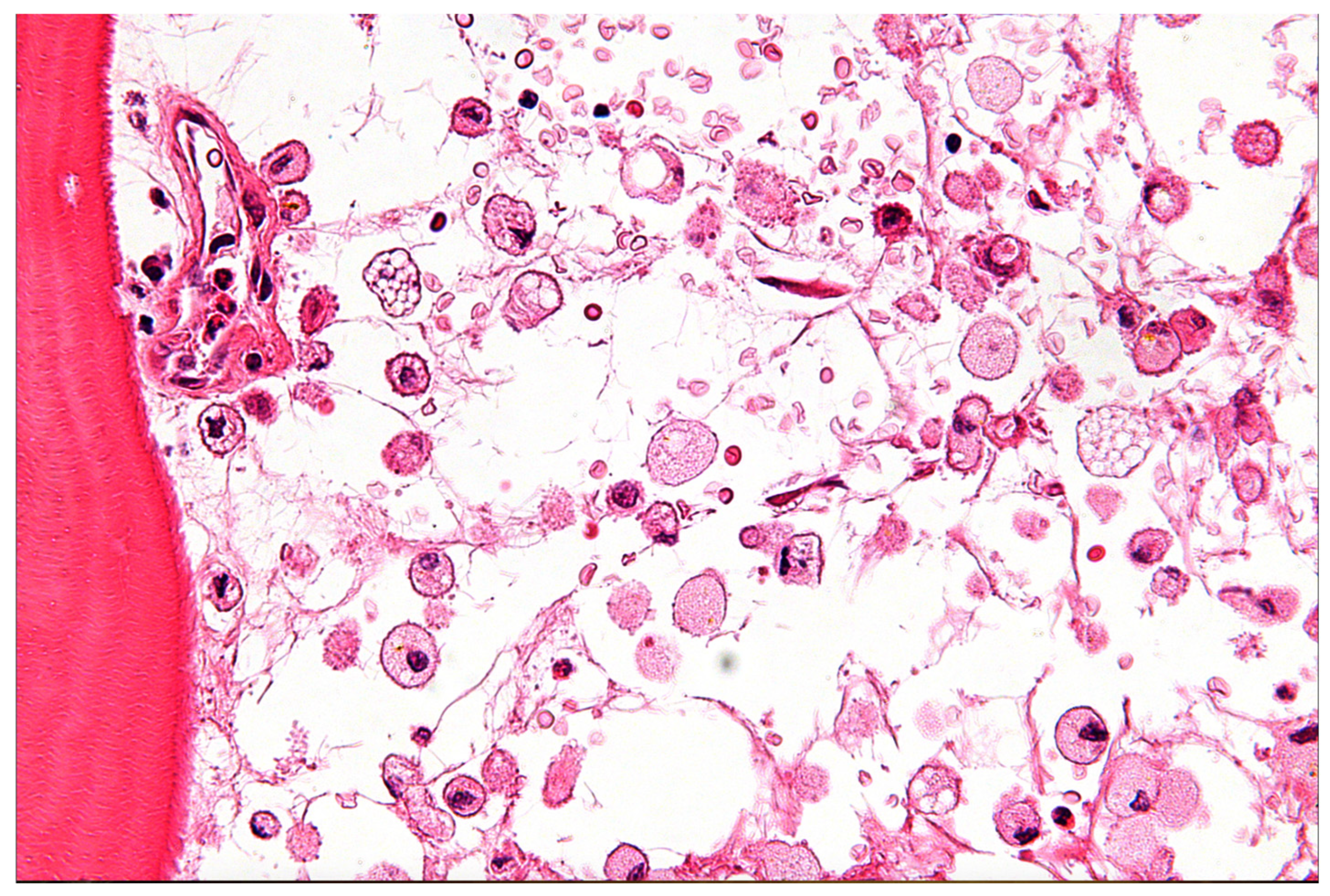
3.2. Lysosomal Storage Diseases (e.g., Niemann–Pick, Gaucher)
3.3. Glucose Metabolism Disorders (e.g., GLUT1 Deficiency, Diabetes-Associated Cognitive Decline)
3.4. Overarching Challenges in the Diagnosis of Neurometabolic Disorders
4. Cerebrospinal Fluid as a Source for Cell-Free DNA Biomarkers in CNS Pathologies
4.1. Unique Characteristics and Functions of Cerebrospinal Fluid
4.2. Advantages of CSF-Derived cfDNA over Plasma cfDNA for CNS Pathologies
5. Epigenetic Profiling of cfDNA: Fundamental Mechanisms and Methodologies
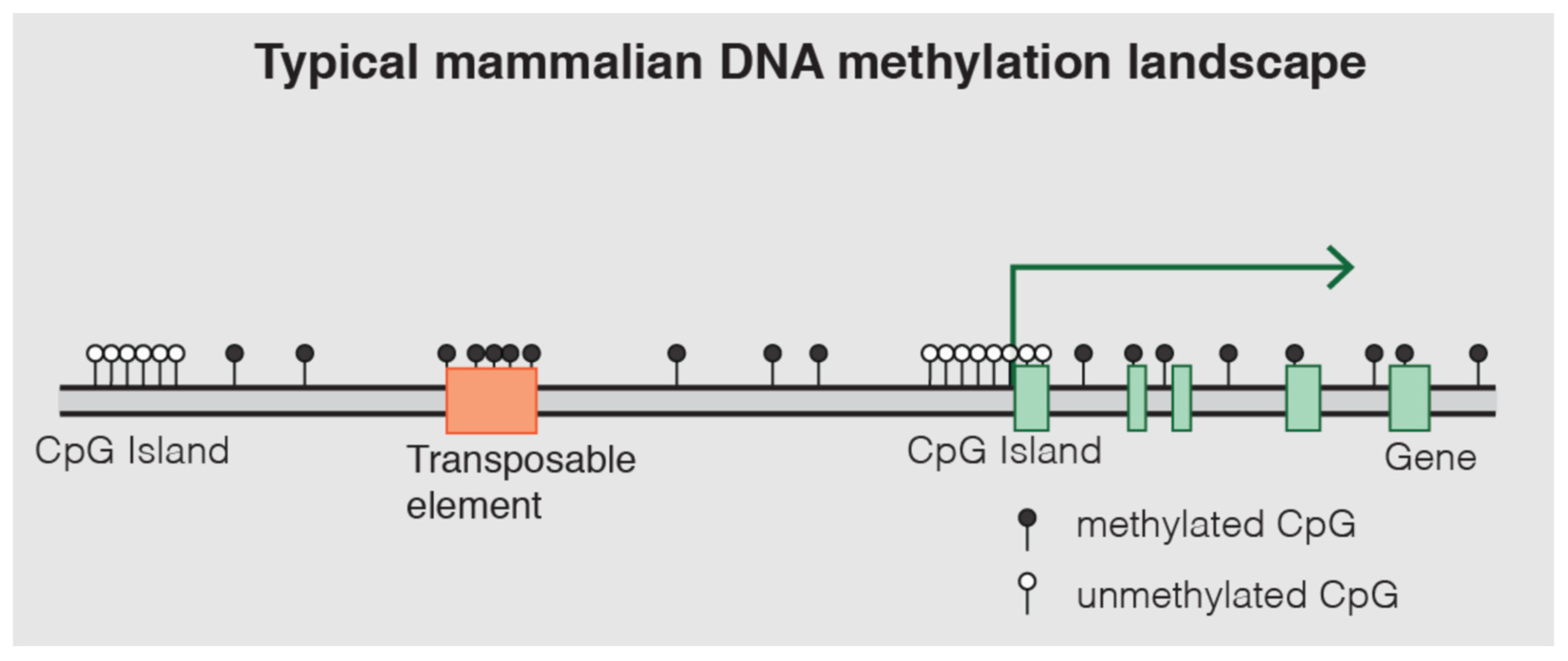
5.1. DNA Methylation: Mechanisms and Role in Gene Regulation
5.2. Methodologies for Methylome Interrogation in cfDNA
5.2.1. cfMeDIP-Seq
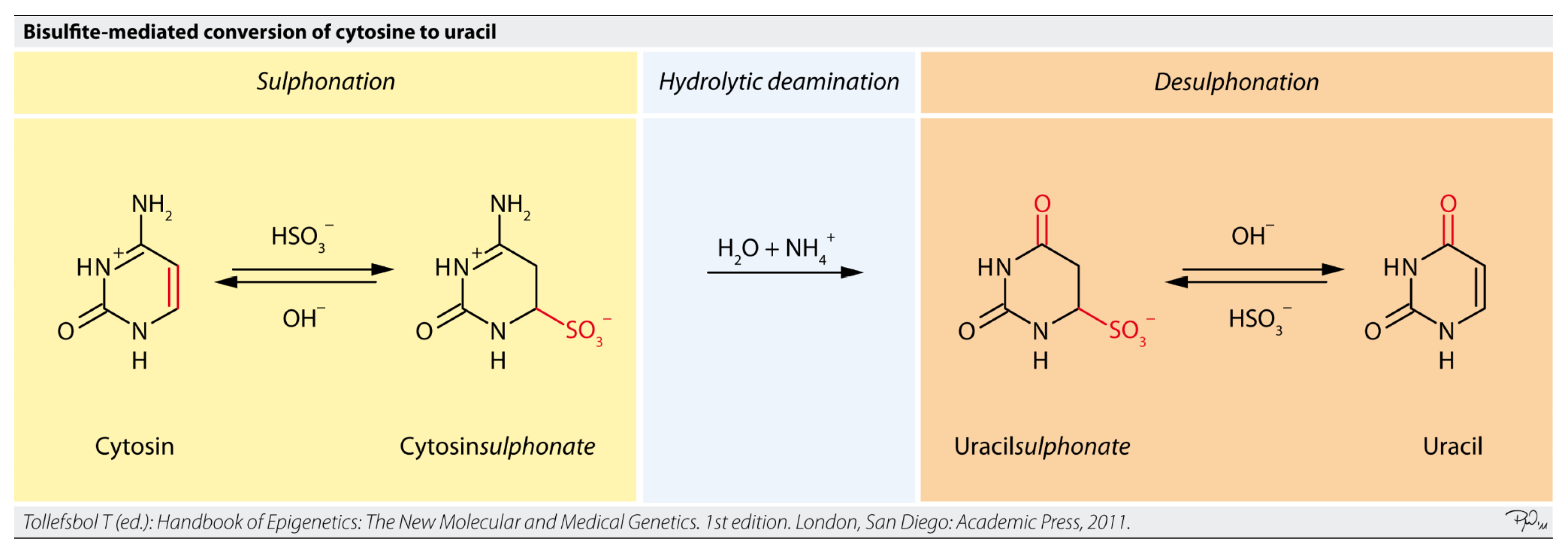
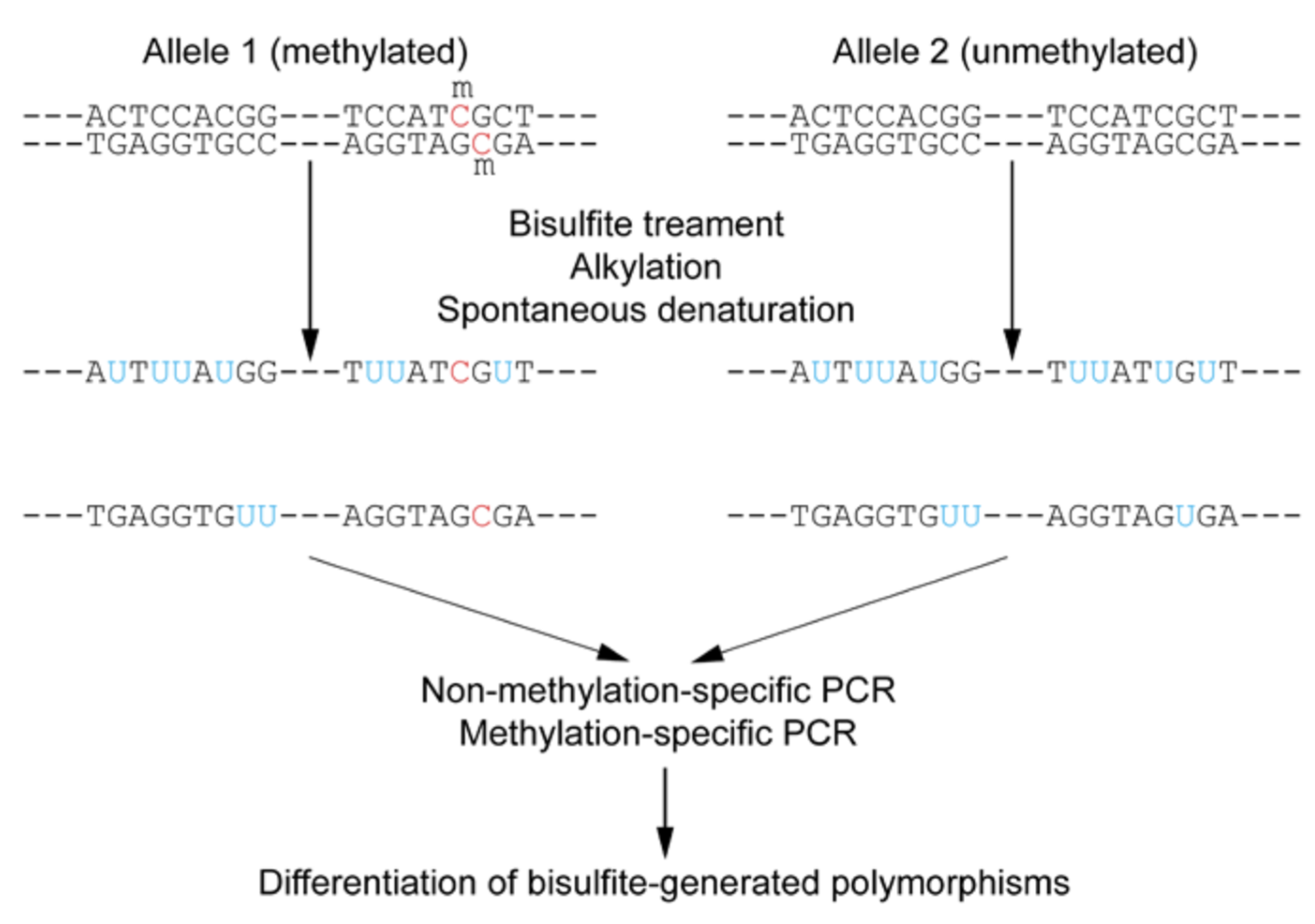
5.2.2. Targeted Bisulfite Sequencing
5.2.3. Enzymatic Methyl-Sequencing (EM-Seq) and EPIC Arrays
5.2.4. Emerging Low-Input DNA Methylation Sequencing Methods
- TAPS (TET-assisted pyridine borane sequencing): This is a bisulfite-free technique that uses enzymatic oxidation and mild chemical reduction to detect 5-methylcytosine (5mC) directly. TAPS avoids the harsh bisulfite treatment, thus preserving DNA integrity and sequence complexity. Only methylated cytosines are converted to thymine, minimizing DNA fragmentation and mapping bias. This yields higher sequencing quality, improved read mapping, and lower cost compared to traditional bisulfite sequencing.
- UBS-seq (Ultra-fast Bisulfite Sequencing): This is an optimized bisulfite sequencing protocol tailored for ultra-low DNA inputs. UBS-seq uses highly concentrated bisulfite and high temperature for a very short duration (~10 min) to achieve complete C → U conversion. The much shorter treatment time dramatically reduces DNA degradation and false conversions, producing ~20-fold lower background noise than conventional bisulfite sequencing. With less bias and damage, UBS-seq enables accurate base-resolution methylation mapping from extremely small DNA amounts (even single cells or just a few nanograms of cfDNA).
- LABS (Linear Amplification-Based Bisulfite Sequencing): LABS is a recently introduced method that replaces PCR with linear pre-amplification to build sequencing libraries from bisulfite-converted DNA. By amplifying DNA fragments in a linear, unbiased manner, LABS avoids the selective loss of rare molecules often seen in exponential (PCR) amplification. LABS has demonstrated improved detection of tumor-specific methylation signals in cfDNA, enhancing sensitivity for liquid biopsies where DNA is scarce.
- Nanopore-based methylation detection: Long-read nanopore sequencing (e.g., Oxford Nanopore Technologies) can directly identify methylated bases on single DNA molecules in real time. As DNA passes through the nanopore, methylation alters the electrical current trace, enabling base-resolution 5mC detection without bisulfite conversion or PCR. This single-molecule approach preserves native DNA (no chemical damage) and simultaneously reads genetic and epigenetic information. Nanopore sequencing has been used to profile methylation in minute cfDNA quantities (on the order of nanograms), successfully detecting differentially methylated regions in low-abundance cell-free samples. Although error rates are higher than short-read methods, ongoing improvements (e.g., higher consensus accuracy and machine learning signal processing) are enhancing its sensitivity and reliability for low-input methylation analysis. Although error rates are higher than short-read methods, ongoing improvements (e.g., higher consensus accuracy and machine learning signal processing) are enhancing its sensitivity and reliability for low-input methylation analysis.
5.3. Other Epigenetic Mechanisms: Histone Modifications and Non-Coding RNAs
5.4. Current Status of DNA Methylation Sequencing Methods
6. Translational Applications: Mapping Brain Cell-Type-Specific Injury, Inflammatory Signaling, and Metabolic Reprogramming
6.1. Brain Cell-Type-Specific Injury
6.2. Inflammatory Signaling
6.3. Metabolic Reprogramming
7. Diagnostic and Prognostic Utility of CSF cfDNA Methylation Biomarkers
7.1. Comparison with Traditional CSF Markers and Neuroimaging
7.2. Diagnostic and Prognostic Applications in Neurometabolic Disorders
- Early Detection and Differential Diagnosis: The capacity of cfDNA methylation to identify specific tissue and cell-type signatures means it can potentially detect early cellular damage or dysfunction in the brain before widespread clinical symptoms emerge [16]. This is particularly critical for MBDs, where early intervention can significantly impact disease progression and patient outcomes [2]. Moreover, by providing highly specific molecular fingerprints, cfDNA methylation patterns can aid in differentiating between various MBDs that share common, non-specific clinical presentations, or distinguish them from other neurological disorders they mimic [11]. For example, in CNS tumors, methylation-based classification of CSF cfDNA has shown high accuracy in discriminating major malignant brain tumor types, approaching the accuracy of standard-of-care tissue biopsies [93]. While MBDs are not tumors, this demonstrates the principle of specific classification.
- Monitoring Disease Progression and Treatment Response: The dynamic nature of cfDNA, with its short half-life, allows for repeated sampling and real-time monitoring of disease activity [9]. Changes in the levels or methylation patterns of cfDNA originating from specific brain cell types could indicate progression of neurodegeneration, shifts in inflammatory states, or the effectiveness of therapeutic interventions [9]. This is valuable for assessing the impact of therapies aimed at correcting metabolic defects or mitigating neuroinflammation. For instance, in glioma, longitudinal CSF cfDNA monitoring has shown changes in tumor-associated variant allele frequencies in response to chemoradiation, even through pseudoprogression [95]. This principle can be extended to MBDs to track the molecular response to treatments.
- Prognostic Insights: Elevated cfDNA concentrations, particularly of circulating tumor DNA (ctDNA) in oncology, often correlate with overall tumor burden and advanced disease stage, indicating poorer clinical outcomes [4]. While direct correlations for MBDs are still emerging, it is plausible that specific cfDNA methylation signatures or quantitative changes could provide prognostic information, predicting disease severity, progression rates, or response to specific therapies. This could enable more precise risk stratification and personalized treatment planning for patients with MBDs (Table 2) [9].
7.3. Comparison of CSF cfDNA Methylation Profiling Vs. Conventional Diagnostic Workups
8. Challenges and Future Directions in Epigenetic Liquid Biopsy for Neurometabolic Disorders
8.1. Technical Challenges
- Low DNA Concentration: While CSF cfDNA concentrations can be higher than plasma for CNS pathologies, the absolute quantities remain low compared to traditional tissue biopsies [5]. This low input can compromise the sensitivity and reliability of methylation assays, increasing the risk of false-positive results [4,67]. Methods like cfMeDIP-seq and enzymatic methyl-sequencing (EM-seq) are designed for low-input samples, but consistent high-quality data remains a challenge [64]. Ultra-low DNA inputs often require additional PCR amplification to generate sequencing libraries, but this step can introduce bias and uneven genomic coverage. When starting material is scarce, certain fragments may be preferentially amplified while others drop out, yielding an incomplete representation of the methylome. These factors together mean that key CpG sites or loci might be missed due to stochastic sampling errors. Approaches such as incorporating unique molecular identifiers and optimizing library preparation protocols (e.g., single-stranded library prep or milder enzymatic conversion) are being explored to mitigate PCR-induced artifacts, but achieving uniform, comprehensive methylation coverage from minimal CSF cfDNA remains challenging
- DNA Integrity and Degradation: Bisulfite conversion, a cornerstone of many methylation analysis methods, can cause significant DNA fragmentation and degradation, impacting sequencing quality and accuracy [4]. While EM-seq offers a milder, less degradative alternative, optimizing library preparation for highly fragmented cfDNA is crucial [67].
- Contamination and Background Noise: Although CSF generally has lower non-tumor cfDNA contamination than plasma, contamination from cellular DNA during sample collection or processing can still occur [3,5]. This can introduce high molecular weight DNA contamination, affecting the accuracy of methylation classification [5]. Careful sample handling protocols, including immediate centrifugation and proper storage, are essential to minimize this [11].
- Limited Cell-of-Origin Resolution: Disease-relevant methylation signals from rare CNS cell populations (e.g., specific neuron or glia subtypes) can be diluted by more abundant DNA from other sources, making it hard to detect subtle, cell-specific patterns. Current deconvolution algorithms can infer cell-type contributions, but their accuracy is constrained by the need for robust reference methylation maps for diverse brain cell types (many of which are still being refined). In practice, this limited resolution means that some epigenetic biomarkers might be obscured by background noise or misattributed, underscoring the technical need for more sensitive methods and comprehensive reference datasets to pinpoint the origins of CSF cfDNA methylation signals.
- Standardization of Assays: A major barrier to clinical adoption is the lack of standardized protocols for cfDNA collection, processing, and analysis [11]. Variability in laboratory procedures, from DNA extraction kits (e.g., MagMAX) to library preparation (e.g., SureSelect XT HS2) and sequencing platforms (e.g., Illumina NovaSeq), can lead to inconsistent results across studies and laboratories [5]. Specifically, differences in CSF sample collection and handling significantly influence cfDNA yield and quality. Delays in processing, suboptimal storage temperatures, or repeated freeze–thaw cycles may degrade cfDNA or introduce cellular DNA contamination, confounding downstream methylation analysis. Additionally, immediate processing (prompt centrifugation, cold storage) and standardized handling are essential, as inconsistent pre-analytical protocols can lead to variability in cfDNA concentrations and fragment profiles. Establishing robust, reproducible, and universally accepted protocols is critical for validating cfDNA methylation biomarkers for routine clinical use [77].
8.2. Interpretive Challenges
- Data Complexity and Bioinformatics: Genome-wide methylation profiling generates vast, high-dimensional datasets that require significant computational resources and specialized bioinformatics expertise for processing, analysis, and interpretation [4]. Developing user-friendly software tools and pipelines that can accurately deconvolve tissue-of-origin signals from complex cfDNA mixtures and identify disease-specific methylation patterns is an ongoing challenge [92].
- Cell-Type Deconvolution Accuracy: While deconvolution algorithms (e.g., CelFiE, CelFEER, cfDecon) are advancing, accurately estimating cell-type proportions from cfDNA methylation data, especially for rare cell types or in the presence of unknown cell types, remains challenging [92]. The accuracy of these methods relies on robust reference methylome datasets for various brain cell types, which are still being developed [17].
- Dynamic Nature of Epigenetic Signatures: Epigenetic signatures can change throughout disease progression or in response to treatment [11]. While this dynamism offers opportunities for monitoring, it also adds complexity to interpretation, as a single methylation profile may not capture the full disease trajectory. This necessitates longitudinal studies and the development of dynamic predictive models.
- Overfitting and Generalizability: Methylation classifiers, especially for rare diseases or subtypes, can suffer from overfitting if training datasets are not comprehensive or representative [61]. This can limit their generalizability to new, unseen patient populations, potentially leading to misclassification [61].
8.3. Clinical Integration and Regulatory Challenges
- Invasiveness of CSF Collection: While less invasive than brain biopsy, lumbar puncture for CSF collection is still an invasive procedure that can cause discomfort and carries risks such as post-dural puncture headache [5]. This limits its applicability for widespread screening or very frequent monitoring, particularly in pediatric populations where ethical standards are stricter [6].
- Regulatory Frameworks: The regulatory landscape for novel liquid biopsy biomarkers, especially for non-oncological CNS conditions, is still evolving [6]. Clear guidelines for assay validation, clinical utility, and reimbursement are necessary to accelerate adoption. The FDA’s emphasis on clear scientific rationale, risk minimization, and comprehensive informed consent for biopsies in clinical trials underscores the stringent requirements for new diagnostic tools [6].
- Cost-Effectiveness: The advanced technologies required for cfDNA methylation analysis, such as next-generation sequencing, can be expensive, with costs primarily driven by consumables [4]. While liquid biopsies can offer downstream economic benefits by avoiding more invasive procedures or guiding more efficient therapy allocation, the initial cost remains a significant barrier to widespread clinical use, particularly for rare diseases [11]. Developing more cost-effective molecular profiling methods and non-proprietary assay panels is crucial [11].
- Ethical Considerations: Beyond the invasiveness of CSF collection, data privacy concerns and ethical issues related to genetic and epigenetic information derived from liquid biopsies need careful consideration, especially as these technologies become more accessible [99].
8.4. Future Directions
- Integration with Artificial Intelligence (AI) and Machine Learning: AI and machine learning algorithms are pivotal for processing complex cfDNA methylation data, improving diagnostic accuracy, and identifying novel methylation signatures [7]. AI can enhance biomarker discovery, optimize classification models, and aid in the interpretation of complex genomic and epigenomic landscapes [91].
- Multi-Omics Approaches: Combining cfDNA methylation profiling with other omics data, such as proteomics (e.g., neurofilament light chain, tau, α-synuclein), transcriptomics (cfRNA), and metabolomics, can provide a more comprehensive and holistic understanding of disease mechanisms and progression [90]. This integrated approach can capture multi-layered genomic and epigenomic information, enhancing diagnostic and prognostic precision [78].
- Longitudinal Studies and Biomarker Validation: Large-scale, prospective longitudinal studies are essential to validate the diagnostic and prognostic utility of CSF cfDNA methylation biomarkers across diverse patient populations and disease stages [18]. Such studies will help establish robust reference ranges, track dynamic changes over time, and correlate molecular findings with clinical outcomes and treatment responses.
- Development of Less Invasive CSF Collection Methods: Research into less invasive or alternative methods for accessing CNS-derived biomarkers, potentially through advanced blood-based approaches that can overcome BBB limitations or novel micro-invasive techniques, could broaden the applicability of these powerful molecular tools.
- Therapeutic Monitoring and Precision Medicine: Given the reversible nature of epigenetic modifications, cfDNA methylation biomarkers hold significant potential for monitoring the efficacy of epigenetic therapies or other targeted interventions in real-time [11]. This aligns with the principles of precision medicine, allowing for tailored therapeutic strategies based on an individual’s unique molecular profile and disease response [4].
9. Conclusions
- Systematic benchmarking of cfDNA epigenetic profiling techniques (e.g., cfMeDIP-seq, EM-seq, bisulfite sequencing) in CSF to determine sensitivity, reproducibility, and cell-type resolution across platforms.
- Longitudinal studies in preclinical models and patient cohorts to map cfDNA methylation dynamics in response to CNS metabolic stress or therapeutic interventions.
- Development of computational frameworks for integrative deconvolution of cfDNA sources, enabling refined tissue- and cell-type attribution within complex CNS pathologies.
- Standardization of protocols for cfDNA extraction, library preparation, and data analysis to enable cross-study comparability and clinical scalability.
- Integration of cfDNA epigenetic profiles with other omics modalities (e.g., proteomics, metabolomics) to create comprehensive disease signatures.
- Development of minimally invasive CSF collection techniques or surrogate biomarkers to enhance patient accessibility and clinical feasibility.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bais, S.; Kumari, R.; Dongre, N.; Panwar, A.S. Insights into neurometabolic diseases. AIMS Mol. Sci. 2024, 11, 116–139. [Google Scholar] [CrossRef]
- Fernández-Eulate, G.; Carreau, C.; Benoist, J.; Lamari, F.; Rucheton, B.; Shor, N.; Nadjar, Y. Diagnostic approach in adult-onset neurometabolic diseases. J. Neurol. Neurosurg. Psychiatry 2022, 93, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, R.; Fujioka, Y.; Hata, N.; Kuga, D.; Hatae, R.; Sangatsuda, Y.; Nakamizo, A.; Mizoguchi, M.; Yoshimoto, K. Liquid Biopsy for Glioma Using Cell-Free DNA in Cerebrospinal Fluid. Cancers 2024, 16, 1009. [Google Scholar] [CrossRef] [PubMed]
- CD Genomics. Cell-free DNA as a Biomarker in Precision Oncology. 2023. Available online: https://www.cd-genomics.com/resource-data-precision-oncology-cfdna.html (accessed on 30 June 2025).
- Cornelli, L.; Van Paemel, R.; Ferro Dos Santos, M.R.; Roelandt, S.; Willems, L.; Vandersteene, J.; Baert, E.; Mus, L.M.; Van Roy, N.; De Wilde, B.; et al. Diagnosis of pediatric central nervous system tumors using methylation profiling of cfDNA from cerebrospinal fluid. Clin. Epigenetics 2024, 16, 87. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- BioIVT. Unpacking the FDA Draft Guidance on Biopsies in Clinical Trials. Available online: https://bioivt.com/blogs/unpacking-the-fda-draft-guidance-on-biopsies-in-clinical-trials (accessed on 30 June 2025).
- Akabane, M.; Imaoka, Y.; Kawashima, J.; Pawlik, T.M. Advancing precision medicine in hepatocellular carcinoma: Current challenges and future directions in liquid biopsy, immune microenvironment, single nucleotide polymorphisms, and conversion therapy. Hepatic Oncol. 2025, 12, 2493457. [Google Scholar] [CrossRef] [PubMed]
- Armakolas, A.; Kotsari, M.; Koskinas, J. Liquid Biopsies, Novel Approaches and Future Directions. Cancers 2023, 15, 1579. [Google Scholar] [CrossRef] [PubMed]
- Aydın, Ş.; Özdemir, S.; Adıgüzel, A. The Potential of cfDNA as Biomarker: Opportunities and Challenges for Neurodegenerative Diseases. J. Mol. Neurosci. 2025, 75, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Life in the Lab Stuff. Cell-Free DNA (cfDNA) vs. Circulating Tumor DNA (ctDNA) Explained. 2025. Available online: https://www.thermofisher.com/blog/life-in-the-lab/cfdna-vs-ctdna/ (accessed on 18 June 2025).
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-free DNA-based liquid biopsies in neurology. Brain 2023, 146, 1758–1774. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, F.; Su, Y.; Liu, X.; Zhao, L.; Wu, Z.; Liu, Y.; Zhang, L. Cell-free DNA: A metabolic byproduct with diagnostic and prognostic potential in rheumatic disorders. Front. Pharmacol. 2025, 16, 1537934. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Tomar, S.; Pathak, S. Epigenetics and Cancer: A Comprehensive Review. Asian Pac. J. Cancer Biol. 2023, 8, 75–89. [Google Scholar] [CrossRef]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532999/ (accessed on 18 June 2025).
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Allen, E.G.; Jin, P. Cell-free DNA methylation as a potential biomarker in brain disorders. Epigenomics 2022, 14, 369–374. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cai, M.; Zhou, J.; McKennan, C.; Wang, J. scMD facilitates cell type deconvolution using single-cell DNA methylation references. Commun. Biol. 2024, 7, 1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McEwen, A.E.; Leary, S.E.S.; Lockwood, C.M. Beyond the Blood: CSF-Derived cfDNA for Diagnosis and Characterization of CNS Tumors. Front. Cell Dev. Biol. 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, J.; Wang, W.; Li, Y. A characteristic comparison of cell-free DNA isolated from plasma and cerebrospinal fluid in patients with cancer. J. Clin. Oncol. 2023, 41, e14008. [Google Scholar] [CrossRef]
- File:EPIC-seq Workflow.png. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:EPIC-seq_workflow.png&oldid=859065986 (accessed on 30 June 2025).
- Pia, S.; Lui, F. Melas Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532959/ (accessed on 18 June 2025).
- File:Mitochondrial Inheritance.svg. Wikimedia Commons. Published 8 March 2022. Available online: https://commons.wikimedia.org/w/index.php?title=File:Mitochondrial_inheritance.svg&oldid=636413970 (accessed on 22 July 2025).
- File:Mitochondrial DNA lg.jpg. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:Mitochondrial_DNA_lg.jpg&oldid=946178322 (accessed on 30 June 2025).
- MedlinePlus. Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes: MedlinePlus Genetics. 2013. Available online: https://medlineplus.gov/genetics/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes/ (accessed on 30 June 2025).
- El-Hattab, A.W.; Almannai, M.; Scaglia, F. MELAS. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2001; pp. 1993–2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1233/ (accessed on 18 June 2025).
- File:Modified Gomori Trichrome Stain Showing Several Ragged Red Fibers.jpg. Wikimedia Commons. 2023. Available online: https://commons.wikimedia.org/w/index.php?title=File:Modified_Gomori_trichrome_stain_showing_several_ragged_red_fibers.jpg&oldid=763868130 (accessed on 30 June 2025).
- Yu, J.; Paterson, C.; Davies, P.; Palmer, J.C.; Higgins, J.P.T.; Kurian, K.M. Evaluating liquid biopsy biomarkers for early detection of brain metastasis: A systematic review. Neuro-Oncol. Pract. 2025, npaf032. [Google Scholar] [CrossRef]
- Liguori, C.; Stefani, A.; Fernandes, M.; Cerroni, R.; Mercuri, N.B.; Pierantozzi, M. Biomarkers of Cerebral Glucose Metabolism and Neurodegeneration in Parkinson’s Disease: A Cerebrospinal Fluid-Based Study. J. Parkinsons Dis. 2022, 12, 537–544. [Google Scholar] [CrossRef] [PubMed]
- BMJ Best Practice. Common Hereditary Lysosomal Storage Diseases—Symptoms, Diagnosis and Treatment|BMJ Best Practice US. 2025. Available online: https://bestpractice.bmj.com/topics/en-us/1021 (accessed on 30 June 2025).
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2017, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Mikosch, P.; Belmatoug, N.; Carubbi, F.; Cox, T.; Goker-Alpan, O.; Kindmark, A.; Mistry, P.; Poll, L.; Weinreb, N.; et al. Gaucher Disease in Bone: From Pathophysiology to Practice. J. Bone Miner. Res. 2019, 34, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M. Niemann-Pick Disease Type C. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2000; pp. 1993–2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1296/ (accessed on 18 June 2025).
- De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome—NORD (National Organization for Rare Disorders). 2024. Available online: https://rarediseases.org/rare-diseases/glucose-transporter-type-1-deficiency-syndrome/ (accessed on 18 June 2025).
- Peters, T.M.A.; Merx, J.; Kooijman, P.C.; Noga, M.; de Boer, S.; van Gemert, L.A.; Salden, G.; Engelke, U.F.; Lefeber, D.J.; van Outersterp, R.E.; et al. Novel CSF biomarkers of GLUT1 deficiency syndrome: Implications beyond the brain’s energy deficit. medRxiv 2022. [Google Scholar] [CrossRef]
- Steele, G.; Klepper, J.; Poduri, A. GLUT1 Deficiency Syndrome (SLC2A1). Epilepsy Foundation. 2021. Available online: https://www.epilepsy.com/causes/metabolic/glut1 (accessed on 30 June 2025).
- Mattox, A.K.; Yan, H.; Bettegowda, C. The potential of cerebrospinal fluid-based liquid biopsy approaches in CNS tumors. Neuro-Oncol. 2019, 21, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bonomi, C.G.; De Lucia, V.; Mascolo, A.P.; Assogna, M.; Motta, C.; Scaricamazza, E.; Sallustio, F.; Mercuri, N.B.; Koch, G.; Martorana, A. Brain energy metabolism and neurodegeneration: Hints from CSF lactate levels in dementias. Neurobiol. Aging 2021, 105, 333–339. [Google Scholar] [CrossRef] [PubMed]
- File:Gaucher Disease—Very High Mag.jpg. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:Gaucher_disease_-_very_high_mag.jpg&oldid=890184076 (accessed on 30 June 2025).
- File:Epigenetic Mechanisms.jpg. Wikimedia Commons. 2022. Available online: https://commons.wikimedia.org/w/index.php?title=File:Epigenetic_mechanisms.jpg&oldid=721049823 (accessed on 30 June 2025).
- Yu, X.; He, H.; Wen, J.; Xu, X.; Ruan, Z.; Hu, R.; Wang, F.; Ju, H. Diabetes-related cognitive impairment: Mechanisms, symptoms, and treatments. Open Med. 2025, 20, 20241091. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, K.; Xiao, Y. Mechanisms of Cognitive Decline in Newly Diagnosed Diabetics: A Review of Pathophysiological Contributions and Intervention Strategies. MEDS Basic Med. 2024, 2, 13–19. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Wen, Z.; Yang, Y.; Bu, T.; Bu, X.; Ni, Q. Cognitive dysfunction in diabetes: Abnormal glucose metabolic regulation in the brain. Front. Endocrinol. 2023, 14, 1192602. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M. Metabolic Alterations Associated to Brain Dysfunction in Diabetes. Aging Dis. 2015, 6, 304–321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abbatecola, A.M.; Arosio, B.; Cerasuolo, M.; Auriemma, M.C.; Di Meo, I.; Langiano, E.; Rizzo, M.R. Common neurodegenerative pathways in brain aging, cognitive decline, type 2 diabetes & metabolic syndrome. J. Gerontol. Geriatr. 2024, 72, 43–49. [Google Scholar] [CrossRef]
- Mayo Clinic Staff. Mild Cognitive Impairment—Symptoms and Causes. 2024. Available online: https://www.mayoclinic.org/diseases-conditions/mild-cognitive-impairment/symptoms-causes/syc-20354578 (accessed on 30 June 2025).
- Summers, K. Study Unlocks How Diabetes Distorts Memory and Reward Processing. UNLV. 2025. Available online: https://www.unlv.edu/news/release/study-unlocks-how-diabetes-distorts-memory-and-reward-processing (accessed on 30 June 2025).
- Bhasin, G.; Flores, E.; Crew, L.A.; Wirt, R.A.; Ortiz, A.A.; Kinney, J.W.; Hyman, J.M. ACC reward location information is carried by hippocampal theta synchrony and suppressed in a Type 2 Diabetes model. J. Neurosci. 2025, e1546242025. [Google Scholar] [CrossRef] [PubMed]
- File:CSF Circulation.png. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:CSF_circulation.png&oldid=859369762 (accessed on 30 June 2025).
- MedlinePlus. Cerebrospinal Fluid (CSF) Analysis: MedlinePlus Lab Test Information. 2025. Available online: https://medlineplus.gov/lab-tests/cerebrospinal-fluid-csf-analysis/ (accessed on 30 June 2025).
- Czarniak, N.; Kamińska, J.; Matowicka-Karna, J.; Koper-Lenkiewicz, O.M. Cerebrospinal Fluid–Basic Concepts Review. Biomedicines 2023, 11, 1461. [Google Scholar] [CrossRef] [PubMed]
- File:CtDNA in circulation.png. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:CtDNA_in_circulation.png&oldid=871699736 (accessed on 30 June 2025).
- File:Blausen 0617 LumbarPuncture.png. Wikimedia Commons. 2023. Available online: https://commons.wikimedia.org/w/index.php?title=File:Blausen_0617_LumbarPuncture.png&oldid=825516397 (accessed on 30 June 2025).
- File:DNAme landscape.png. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:DNAme_landscape.png&oldid=873650162 (accessed on 30 June 2025).
- Lossi, L.; Castagna, C.; Merighi, A. An Overview of the Epigenetic Modifications in the Brain under Normal and Pathological Conditions. Int. J. Mol. Sci. 2024, 25, 3881. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Younesian, S.; Yousefi, A.M.; Momeny, M.; Ghaffari, S.H.; Bashash, D. The DNA Methylation in Neurological Diseases. Cells 2022, 11, 3439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lio, C.W.J.; Rao, A. TET Enzymes and 5hmC in Adaptive and Innate Immune Systems. Front. Immunol. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Helin, K. DNA methylation: TET proteins—Guardians of CpG islands? EMBO Rep. 2011, 13, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Epigenet. Dev. Dis. 2013, 61, 289–317. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hiemenz, M.; Chang, J. DNA Methylation to the Rescue: An Update for Pathologists Navigating the Future of Precision Tumor Diagnostics. College of American Pathologists. 2025. Available online: https://www.cap.org/member-resources/articles/dna-methylation-to-the-rescue-an-update-for-pathologists-navigating-the-future-of-precision-tumor-diagnostics (accessed on 30 June 2025).
- Kakodkar, P.; Conway, K.; Santana-Santos, L.; McCord, M.; Sukhanova, M.; Castellani, R.; Jamshidi, P. DNA methylation-based classification of the central nervous system tumors, achievements, and challenges. In Advanced Concepts and Strategies in Central Nervous System Tumors; IntechOpen: London, UK, 2025. [Google Scholar] [CrossRef]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, Z.; Kirsh, B.O.; Luo, P.; Pedersen, S.; Bucur, R.C.; Rukavina, N.A.; Bruce, J.P.; Danesh, A.; Riverin, M.; et al. Plasma cell-free DNA methylomes for hepatocellular carcinoma detection and monitoring after liver resection or transplantation. medRxiv 2024. [Google Scholar] [CrossRef]
- Wilson, S.L.; Shen, S.Y.; Harmon, L.; Burgener, J.M.; Triche, T.; Bratman, S.V.; De Carvalho, D.D.; Hoffman, M.M. Sensitive and reproducible cell-free methylome quantification with synthetic spike-in controls. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Biorad. Epigenetics and Chromatin Structure|Bio-Rad. Available online: https://www.bio-rad.com/en-us/applications-technologies/epigenetics-chromatin-structure?ID=LUSNMFHYP (accessed on 30 June 2025).
- CD Genomics. Innovation in WGBS: New Methods for DNA Methylation Analysis. 2023. Available online: https://www.cd-genomics.com/epigenetics/resource-new-wgbs-technology-pbat-wgbs.html (accessed on 30 June 2025).
- File:Bisulfite-Reaction.png. Wikimedia Commons. 2023. Available online: https://commons.wikimedia.org/w/index.php?title=File:Bisulfite-reaction.png&oldid=824921164 (accessed on 30 June 2025).
- File:Wiki Bisulfite Sequencing Figure 1 Small.png. Wikipedia. 2012. Available online: https://en.wikipedia.org/wiki/File:Wiki_Bisulfite_sequencing_Figure_1_small.png (accessed on 30 June 2025).
- Abcam. Bisulfite Sequencing in DNA Methylation Analysis|Abcam. 2017. Available online: https://www.abcam.com/en-us/knowledge-center/dna-and-rna/bisulfite-sequencing (accessed on 30 June 2025).
- De Koker, A.; Raman, L.; Van Paemel, R.; Van der Linden, M.; Van de Velde, S.; Van der Leest, P.; Van Gaever, B.; Zaka, A.; Vermaelen, K.; Demedts, I.; et al. Development of circulating cell-free DNA reduced representation bisulfite sequencing for clinical methylomics diagnostics. bioRxiv 2025. [Google Scholar] [CrossRef]
- What Are the Limitations of Reduced Representation Bisulfite Sequencing (RRBS)?|AAT Bioquest. 2024. Available online: https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-limitations-of-reduced-representation-bisulfite-sequencing-rrbs (accessed on 30 June 2025).
- CD Genomics. DNA Methylation Array Workflow & Analysis: Process Optimization and Data Insights. 2021. Available online: https://www.cd-genomics.com/epigenetics/resource-dna-methylation-array-workflow-optimization-analysis.html (accessed on 30 June 2025).
- CD Genomics. Overview of EM-seq: A New Detection Approach for DNA Methylation—CD Genomics. 2021. Available online: https://www.cd-genomics.com/epigenetics/resource-dna-methylation-detection-em-seq.html (accessed on 30 June 2025).
- Illumina. Infinium MethylationEPIC Kit|Methylation Profiling Array for EWAS. 2019. Available online: https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html (accessed on 30 June 2025).
- Sebastian, A.; Silvy, M.; Coiffard, B.; Reynaud-Gaubert, M.; Magdinier, F.; Chiaroni, J.; Picard, C.; Pedini, P. A review of cell-free DNA and epigenetics for non-invasive diagnosis in solid organ transplantation. Front. Transplant. 2024, 3, 1474920. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tan, W.Y.; Nagabhyrava, S.; Ang-Olson, O.; Das, P.; Ladel, L.; Sailo, B.; He, L.; Sharma, A.; Ahuja, N. Translation of Epigenetics in Cell-Free DNA Liquid Biopsy Technology and Precision Oncology. Curr. Issues Mol. Biol. 2024, 46, 6533–6565. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.A.; Saito, E.R.; Burns, J.M.; Hill, J.T.; Jenkins, T.G. Considering Biomarkers of Neurodegeneration in Alzheimer’s Disease: The Potential of Circulating Cell-Free DNA in Precision Neurology. J. Pers. Med. 2024, 14, 1104. [Google Scholar] [CrossRef] [PubMed]
- Southwood, D.; Singh, S.; Chatterton, Z. Brain-derived cell-free DNA. Neural Regen. Res. 2022, 17, 2213–2214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, Y.; Conneely, K.N.; Ma, W.; Naviaux, R.K.; Siddique, T.; Allen, E.G.; Guingrich, S.; Pascuzzi, R.M.; Jin, P. Whole-genome bisulfite sequencing of cell-free DNA unveils age-dependent and ALS-associated methylation alterations. Cell Biosci. 2025, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Rados, M.; Klotz, S.; Regelsberger, G.; Stögmann, E.; Landegger, L.D.; Fetahu, I.S. Distinct cerebrospinal fluid DNA methylation signatures linked to Alzheimer’s disease. bioRxiv 2025. [Google Scholar] [CrossRef]
- Giannini, L.A.A.; Boers, R.G.; van der Ende, E.L.; Poos, J.M.; Jiskoot, L.C.; Boers, J.B.; van IJcken, W.F.J.; Dopper, E.G.; Pijnenburg, Y.A.L.; Seelaar, H.; et al. Distinctive cell-free DNA methylation characterizes presymptomatic genetic frontotemporal dementia. Ann. Clin. Transl. Neurol. 2024, 11, 744–756. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, Y.; Wang, W.; Liu, S.; Qiao, X.; Xing, Y.; Zhou, Q.; Zhang, Z. Epigenetic Regulation of Neuroinflammation in Alzheimer’s Disease. Cells 2023, 13, 79. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, I.; Keyser, C.; Zhang, Z.; Rosolia, B.; Hwang, J.-Y.; Zukin, R.S.; Yan, J. Epigenetic regulation of autophagy in neuroinflammation and synaptic plasticity. Front. Immunol. 2024, 15, 1322842. [Google Scholar] [CrossRef] [PubMed]
- File:Blausen 0870 TypesofNeuroglia.png. Wikimedia Commons. 2024. Available online: https://commons.wikimedia.org/w/index.php?title=File:Blausen_0870_TypesofNeuroglia.png&oldid=859366013 (accessed on 1 July 2025).
- Cologna, S.M.; Cluzeau, C.V.; Yanjanin, N.M.; Blank, P.S.; Dail, M.K.; Siebel, S.; Toth, C.L.; Wassif, C.A.; Lieberman, A.P.; Porter, F.D. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 2014, 37, 83–92. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boddupalli, C.S.; Nair, S.; Belinsky, G.; Gans, J.; Teeple, E.; Nguyen, T.H.; Mehta, S.; Guo, L.; Kramer, M.L.; Ruan, J.; et al. Neuroinflammation in neuronopathic Gaucher disease: Role of microglia and NK cells, biomarkers, and response to substrate reduction therapy. elife 2022, 11, e79830. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fawal, M.A.; Davy, A. Impact of Metabolic Pathways and Epigenetics on Neural Stem Cells. Epigenet. Insights 2018, 11, 2516865718820946. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peters, T.M.A.; Merx, J.; Kooijman, P.C.; Noga, M.; de Boer, S.; van Gemert, L.A.; Salden, G.; Engelke, U.F.H.; Lefeber, D.J.; van Outersterp, R.E.; et al. Novel cerebrospinal fluid biomarkers of glucose transporter type 1 deficiency syndrome: Implications beyond the brain’s energy deficit. J. Inherit. Metab. Dis. 2023, 46, 66–75. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Laufer, B.I.; Hasegawa, Y.; Zhang, Z.; Hogrefe, C.E.; Del Rosso, L.A.; Haapanen, L.; Hwang, H.; Bauman, M.D.; Van de Water, J.; Taha, A.Y.; et al. Multi-omic brain and behavioral correlates of cell-free fetal DNA methylation in macaque maternal obesity models. Nat. Commun. 2022, 13, 5538. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Myrou, A.; Barmpagiannos, K.; Ioakimidou, A.; Savopoulos, C. Molecular Biomarkers in Neurological Diseases: Advances in Diagnosis and Prognosis. Int. J. Mol. Sci. 2025, 26, 2231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Li, J.; Yang, S.; Huang, Y.; Liu, X.; Fan, Y.; King, I.; Li, Y.; Li, Y. cfDecon: Accurate and Interpretable methylation-based cell type deconvolution for cell-free DNA. bioRxiv 2025. [Google Scholar] [CrossRef]
- Zuccato, J.A.; Patil, V.; Mansouri, S.; Voisin, M.; Chakravarthy, A.; Shen, S.Y.; Nassiri, F.; Mikolajewicz, N.; Trifoi, M.; Skakodub, A.; et al. Cerebrospinal fluid methylome-based liquid biopsies for accurate malignant brain neoplasm classification. Neuro-Oncol. 2023, 25, 1452–1460. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riviere-Cazaux, C.; Dong, X.; Mo, W.; Kumar, R.; Dai, C.; Carlstrom, P.L.; Munoz-Casabella, A.; Ghadimi, K.; Nesvick, L.C.; Andersen, M.K.; et al. Longitudinal Glioma Monitoring via Cerebrospinal Fluid Cell-Free DNA. Clin. Cancer Res. 2025, 31, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L. Cell-Free DNA Methylation Profiling Analysis-Technologies and Bioinformatics. Cancers 2019, 11, 1741. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iser, F.; Hinz, F.; Hoffmann, D.C.; Grassl, N.; Güngoör, C.; Meyer, J.; Dörner, L.; Hofmann, L.; Kelbch, V.; Göbel, K.; et al. Cerebrospinal Fluid cfDNA Sequencing for Classification of Central Nervous System Glioma. Clin. Cancer Res. 2024, 30, 2974–2985. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, R.; Li, S.; Stackpole, M.L.; Li, Q.; Zhou, X.J.; Li, W. cfTools: An R/Bioconductor package for deconvolving cell-free DNA via methylation analysis. Bioinform. Adv. 2025, 5, vbaf108. [Google Scholar] [CrossRef] [PubMed]
- Keukeleire, P.; Makrodimitris, S.; Reinders, M. Cell type deconvolution of methylated cell-free DNA at the resolution of individual reads. NAR Genom. Bioinform. 2023, 5, lqad048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Research and Markets. Liquid Biopsy Market Forecast to Reach US$13.44 Billion by 2030 at 12.19% CAGR|Next-Gen Sequencing and PCR Breakthroughs Fuel Innovation in Liquid Biopsy Diagnostics. GlobeNewswire News Room. 2025. Available online: https://www.globenewswire.com/news-release/2025/04/09/3058183/28124/en/Liquid-Biopsy-Market-Forecast-to-Reach-US-13-44-Billion-by-2030-at-12-19-CAGR-Next-Gen-Sequencing-and-PCR-Breakthroughs-Fuel-Innovation-in-Liquid-Biopsy-Diagnostics.html (accessed on 30 June 2025).
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflamm. 2016, 13, 36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Magner, M.; Szentiványi, K.; Svandová, I.; Ješina, P.; Tesařová, M.; Honzík, T.; Zeman, J. Elevated CSF-lactate is a reliable marker of mitochondrial disorders in children even after brief seizures. Eur. J. Paediatr. Neurol. 2011, 15, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Fujimura, H.; Nishikawa, Y.; Yorifuji, S.; Mezaki, T.; Hirono, N.; Nishitani, N.; Kameyama, M. Marked reduction in CSF lactate and pyruvate levels after CoQ therapy in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Acta Neurol. Scand. 1991, 83, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jiang, J.; Chang, L.; Zhang, B.; Zhu, X.; Niu, F. Multisystem clinicopathologic and genetic analysis of MELAS. Orphanet J. Rare Dis. 2024, 19, 487. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Toribe, Y.; Yanagihara, K.; Mano, T.; Akagi, M.; Suzuki, Y. Diagnostic accuracy of blood and CSF lactate in identifying children with mitochondrial diseases affecting the central nervous system. Brain Dev. 2012, 34, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Molina, M.P.; Bernabeu-Sanz, Á.; Ramos-González, A.; Morales-Conejo, M.; Delmiro, A.; Domínguez-González, C.; Arenas, J.; Martín, M.A.; González de la Aleja, J. Magnetic resonance spectroscopy in MELAS syndrome: Correlation with CSF and plasma metabolite levels and change after glutamine supplementation. Neuroradiology 2024, 66, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Natsume, J.; Ishihara, N.; Azuma, Y.; Nakata, T.; Takeuchi, T.; Tanaka, M.; Sakaguchi, Y.; Okai, Y.; Ito, Y.; Yamamoto, H.; et al. Lenticular nuclei to thalamic ratio on PET is useful for diagnosis of GLUT1 deficiency syndrome. Brain Dev. 2021, 43, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Kocabas, A.; Rodriguez-Navas, C.; Miloushev, V.Z.; González-Rodríguez, M.; Lees, H.; Henry, K.E.; Vaynshteyn, J.; Longo, V.; Deh, K.; et al. Imaging brain glucose metabolism in vivo reveals propionate as a major anaplerotic substrate in pyruvate dehydrogenase deficiency. Cell Metab. 2024, 36, 1394–1410.e12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meyer, P.T.; Frings, L.; Rücker, G.; Hellwig, S. 18F-FDG PET in Parkinsonism: Differential Diagnosis and Cognitive Impairment in Parkinson’s disease. J. Nucl. Med. 2017, 58, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Probasco, J.C.; Solnes, L.; Nalluri, A.; Cohen, J.; Jones, K.M.; Zan, E.; Javadi, M.S.; Venkatesan, A. Abnormal brain metabolism on FDG-PET/CT is a common early finding in autoimmune encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e352. [Google Scholar] [CrossRef] [PubMed]
- Akman, C.I.; Provenzano, F.; Wang, D.; Engelstad, K.; Hinton, V.; Yu, J.; Tikofsky, R.; Ichese, M.; De Vivo, D.C. Topography of brain glucose hypometabolism and epileptic network in glucose transporter 1 deficiency. Epilepsy Res. 2015, 110, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, G.; Pinelli, M.; Alagia, M.; Donti, T.; Day-Salvatore, D.L.; Veggiotti, P.; De Giorgis, V.; Lunghi, S.; Vari, M.S.; Striano, P.; et al. Biochemical phenotyping unravels novel metabolic abnormalities and potential biomarkers associated with treatment of GLUT1 deficiency with ketogenic diet. PLoS ONE 2017, 12, e0184022. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, L.; McClain, L.; Shaw, P.; Donnellan, N.; Chu, T.; Finegold, D.; Peters, D. Non-invasive epigenomic molecular phenotyping of the human brain via liquid biopsy of cerebrospinal fluid and next generation sequencing. Eur. J. Neurosci. 2020, 52, 4536–4545. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, R.Y.; Guan, F.; Yuan, Z. Lactate metabolism in neurodegenerative diseases. Neural Regen. Res. 2024, 19, 69–74. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Otto, C.; Kalantzis, R.; Kübler-Weller, D.; Kühn, A.A.; Böld, T.; Regler, A.; Strathmeyer, S.; Wittmann, J.; Ruprecht, K.; Heelemann, S. Comprehensive analysis of the cerebrospinal fluid and serum metabolome in neurological diseases. J. Neuroinflamm. 2024, 21, 234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mastrangelo, M.; Manti, F.; Ricciardi, G.; Cinnante, E.M.C.; Cameli, N.; Beatrice, A.; Tolve, M.; Pisani, F. The diagnostic and prognostic role of cerebrospinal fluid biomarkers in glucose transporter 1 deficiency: A systematic review. Eur. J. Pediatr. 2024, 183, 3665–3678. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Wevers, R.A.; Kamsteeg, E.J.; Scheffer, H.; Verbeek, M.M.; Willemsen, M.A. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: A systematic review. JAMA Neurol. 2013, 70, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
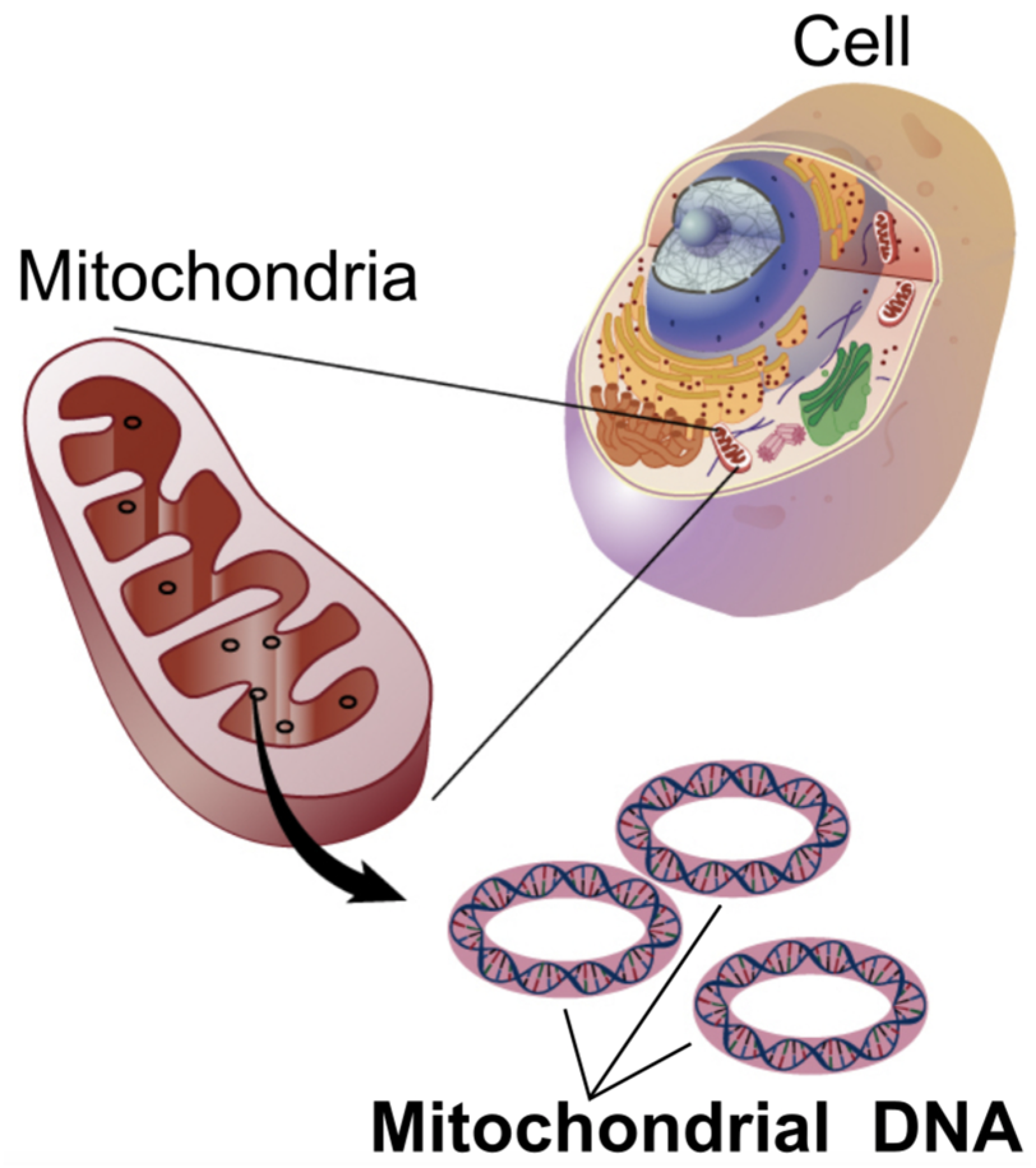
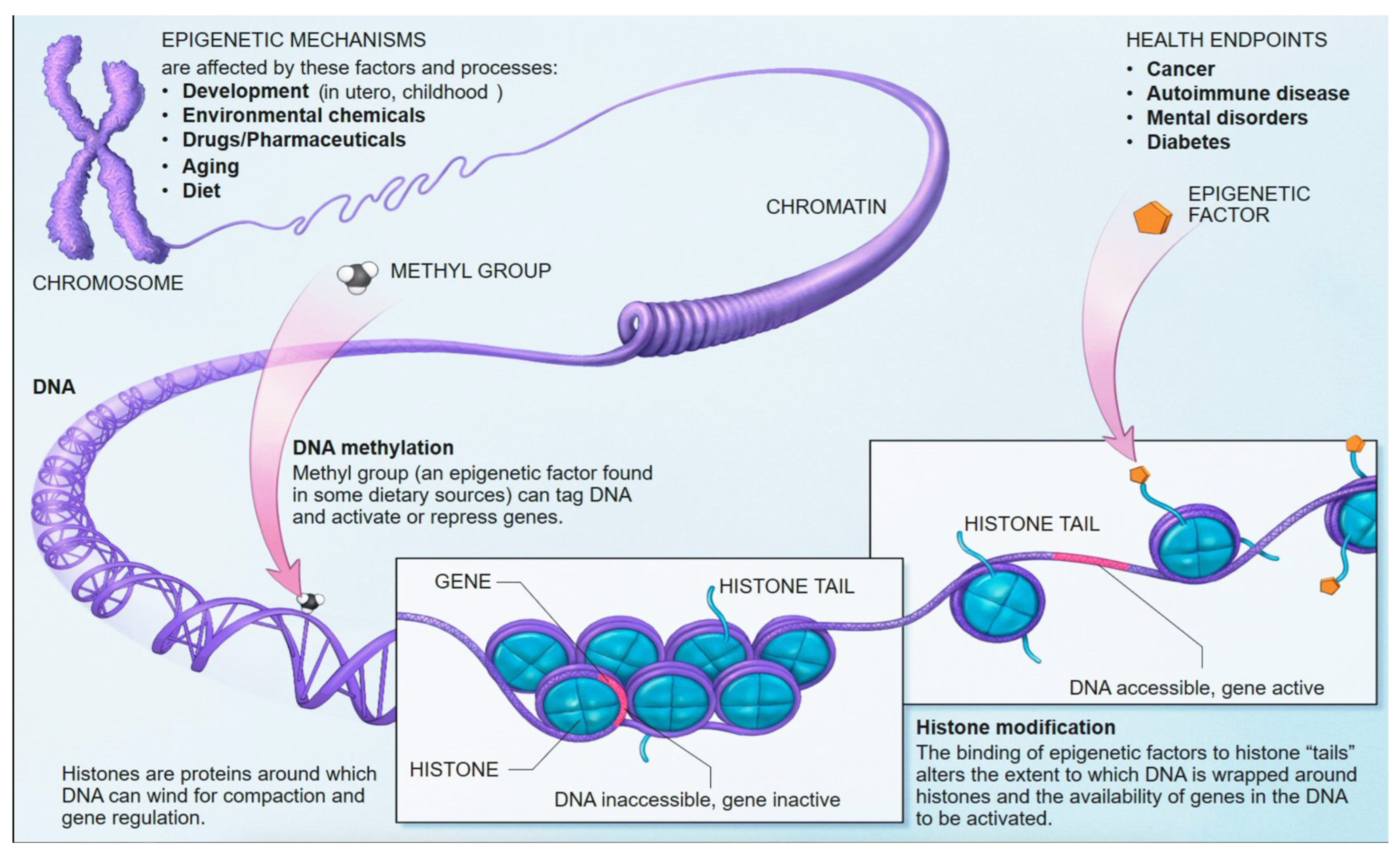
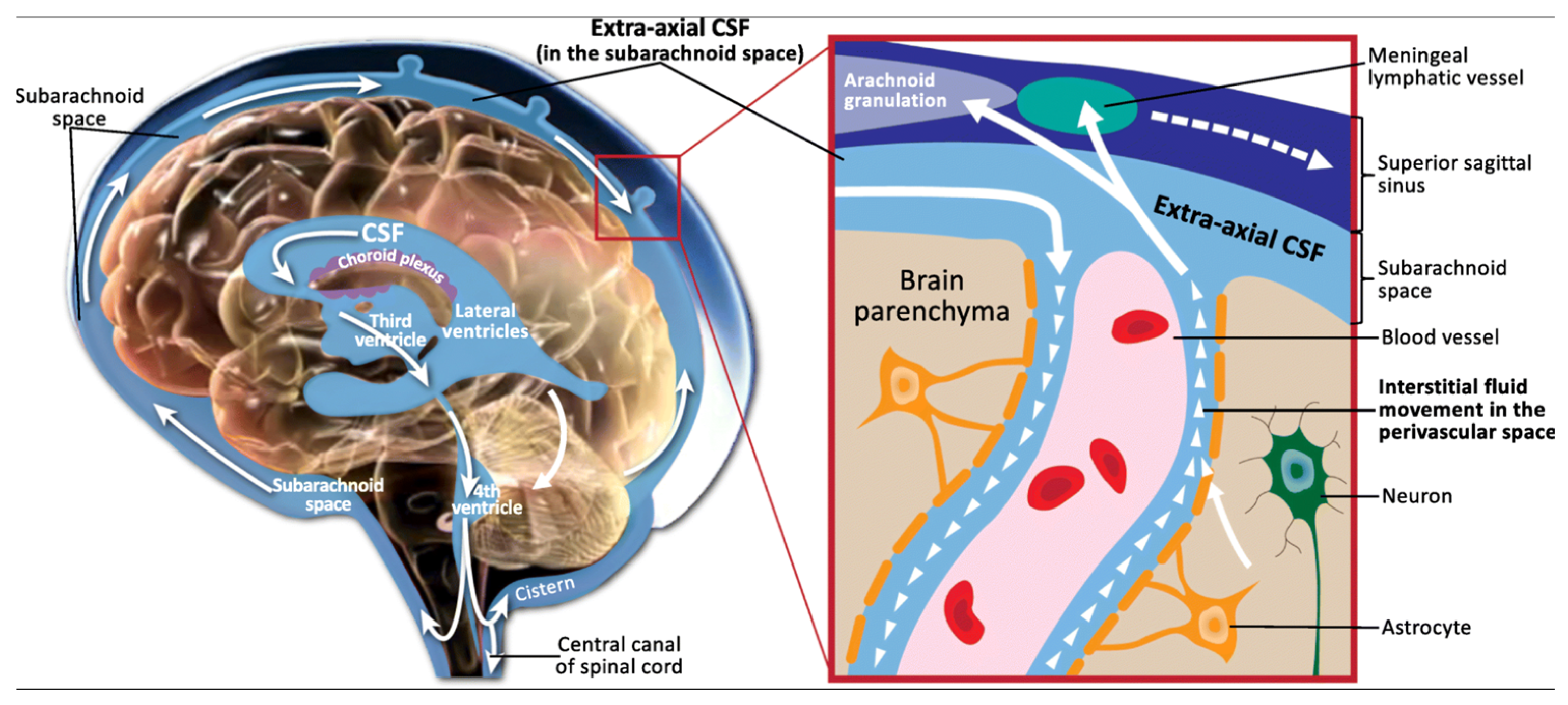
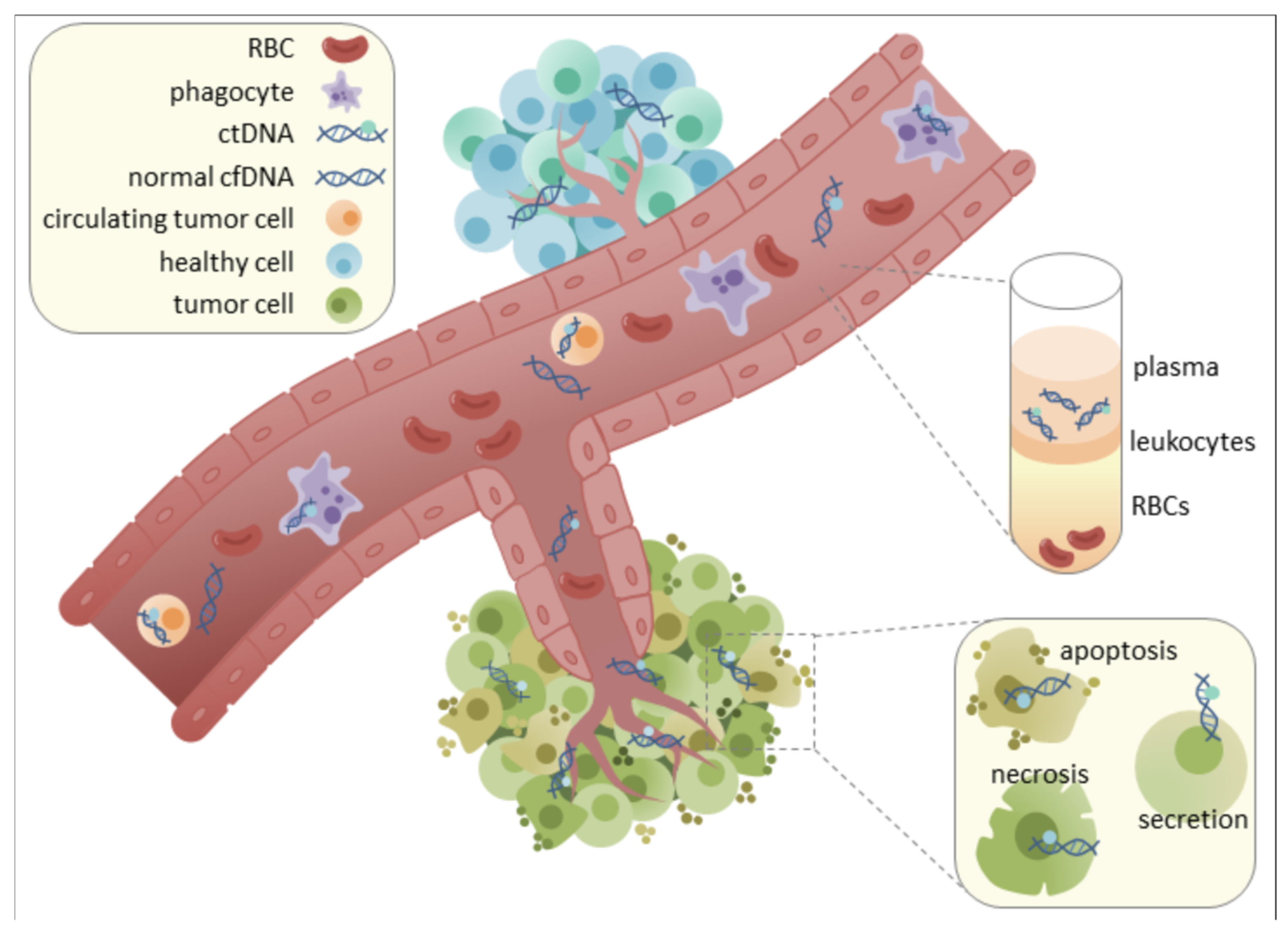
| Method | Sensitivity/Coverage | Relative Cost | Resolution | Min DNA Input |
|---|---|---|---|---|
| cfMeDIP-seq | Moderate genome-wide signal; efficient enrichment of methylated CpGs; good fragment capture in cfDNA3 | Moderate | Regional (~100–200 bp) | ~10 ng |
| Targeted Bisulfite-Seq | High in chosen loci; poor coverage elsewhere due to bisulfite damage1 | Low–Moderate | Base-pair | 5–10 ng |
| RRBS | Enriched CpG islands, lower genome coverage; high damage limits input3 | Low | Base-pair | ~10 ng |
| WGBS | Gold-standard complete methylome; high-depth increases costs | High | Base-pair | ≥100 ng |
| EM-seq | High conversion accuracy, minimal degradation with low input4 | Moderate–High | Base-pair | ~100 pg |
| EPIC Array | Quantitative at defined CpGs only | Low | Probe-level (CpG sites) | ≥200 ng |
| TAPS | Minimal DNA damage; preserves fragment integrity; ≥10 ng cfDNA usage in plasma cfDNA5 | Moderate | Base-pair | ~10–20 ng |
| UBS-seq | ~13× faster bisulfite reaction; lower DNA damage and background noise in cfDNAs6 | Moderate | Base-pair | 1–100 cells (~1–10 ng) |
| LABS | Linear amplification preserves rare cfDNA fragments; single-base genome-wide data from <10 pg7 | Moderate–High | Base-pair | <10 pg (~sub-nanogram) |
| Nanopore Methylation | Single-molecule; long reads; still modest per-site accuracy (~90–95%)8 | Moderate | Base-pair and long-range | ~10 ng |
| Modality | Measured Target | Quantitative/Qualitative Output | Diagnostic Accuracy | Limitations |
|---|---|---|---|---|
| CSF Lactate [100,101,102,103,104] | Anaerobic CNS metabolism | 4.4 mmol/L in MELAS vs. ~1.6 in controls; cut-off > 2.2 mmol/L; AUC 0.994 | High sensitivity (94–100%) and specificity (100%) in pediatric series | Requires lumbar puncture; affected by seizures/infection |
| 1H-MRS (MELAS) [105] | Brain lactate peak (lactate/Cr) | 0.40 ± 0.05 (frontal), 0.32 ± 0.03 (occipital) vs. zero controls; correlated with CSF lactate (r = 0.85) | Qualitative, binary presence detection | Not quantitative; low dynamic range |
| 31P-MRS (Muscle MELAS) [105] | Pi/PCr ratio | Elevated post-exercise; lowered post-recovery | Reflects peripheral mitochondrial dysfunction | Indirect CNS measure; muscle-specific |
| MRI (MELAS) [105] | Stroke-like lesions | FLAIR/T2 hyperintensities, inverted lactate double peak, NAA/Cr low (~0.79 vs. 1.8–2.2) | Visual identification of lesions | Structural, post-lesion; no early detection |
| PET/CSF in Alzheimer’s | Aβ + tau PET vs. CSF Aβ42/t-tau | PET AUC 0.92–0.93; CSF Aβ42/t-tau AUC 0.93–0.94; Sens 97%, Spec 83% | Equally accurate; CSF slightly better | Both require LP or PET; cost |
| GLUT1 Deficiency Syndrome | CSF glucose and CSF/serum ratio | CSF glucose: 34–44% of blood; CSF/serum ratio ~0.34–0.44; no standardized lactate cut-off but often low-normal. | FDG-PET: Lenticular/thalami SUV ratio cut-off ≥ 1.54 discriminates GLUT1-DS vs. epilepsy controls with 100% sensitivity and 98% specificity. Widespread hypometabolism in thalamus, cerebellum, and temporal cortex by SPM | CSF glucose remains a more direct, earlier marker; PET shows high accuracy but requires high-cost imaging |
| Disease | Proposed cfDNA Epigenetic Features | Potential Advantages |
|---|---|---|
| MELAS [37,111,112,113] | Neuron/glia-specific methylation shifts affecting mitochondrial genes | Early detection of cell-type-specific injury; more precise than lactate |
| Lysosomal Disorders (e.g., Niemann-Pick, Gaucher) [114] | Methylation alterations in microglia/neuron cfDNA tied to inflammation and lipid pathways | Better detection of neuroinflammation vs. unstable small molecules |
| GLUT1DS [35,89,111,115,116] | cfDNA methylation changes in glucose transport/metabolism genes (e.g., SLC2A1 targets) | Detects CNS metabolic stress even if glucose is near-normal; complements new metabolic markers |
| Neurodegenerative-Metabolic (e.g., PD, AD, MS) [28,38,100] | cfDNA profiles reflecting insulin signaling, oxidative stress, and synaptic integrity | Enables differentiation from pure neurodegeneration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sporn, K.; Kumar, R.; Marla, K.; Ravi, P.; Vaja, S.; Paladugu, P.; Zaman, N.; Tavakkoli, A. Epigenetic Profiling of Cell-Free DNA in Cerebrospinal Fluid: A Novel Biomarker Approach for Metabolic Brain Diseases. Life 2025, 15, 1181. https://doi.org/10.3390/life15081181
Sporn K, Kumar R, Marla K, Ravi P, Vaja S, Paladugu P, Zaman N, Tavakkoli A. Epigenetic Profiling of Cell-Free DNA in Cerebrospinal Fluid: A Novel Biomarker Approach for Metabolic Brain Diseases. Life. 2025; 15(8):1181. https://doi.org/10.3390/life15081181
Chicago/Turabian StyleSporn, Kyle, Rahul Kumar, Kiran Marla, Puja Ravi, Swapna Vaja, Phani Paladugu, Nasif Zaman, and Alireza Tavakkoli. 2025. "Epigenetic Profiling of Cell-Free DNA in Cerebrospinal Fluid: A Novel Biomarker Approach for Metabolic Brain Diseases" Life 15, no. 8: 1181. https://doi.org/10.3390/life15081181
APA StyleSporn, K., Kumar, R., Marla, K., Ravi, P., Vaja, S., Paladugu, P., Zaman, N., & Tavakkoli, A. (2025). Epigenetic Profiling of Cell-Free DNA in Cerebrospinal Fluid: A Novel Biomarker Approach for Metabolic Brain Diseases. Life, 15(8), 1181. https://doi.org/10.3390/life15081181