
PBPK Modeling of Acetaminophen in Pediatric Populations: Incorporation of SULT Enzyme Ontogeny to Predict Age-Dependent Metabolism and Systemic Exposure


Abstract
1. Introduction
2. Materials and Methods
2.1. Mathematical Characterization of SULT Ontogeny
2.2. Physiologically-Based Pharmacokinetic (PBPK) Modeling
2.3. Sensitivity Analysis
3. Results
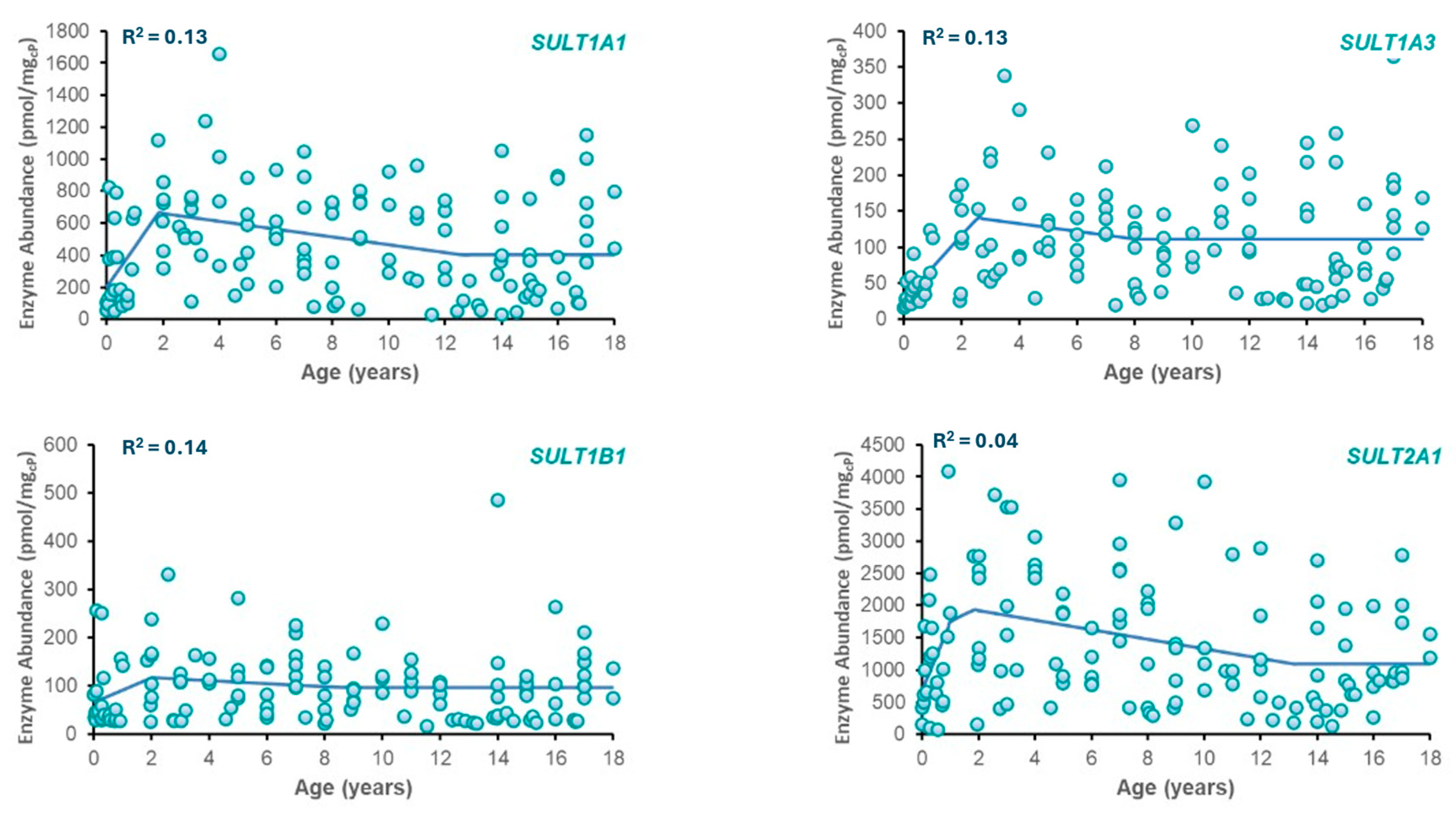
3.1. SULT Ontogeny Modeling
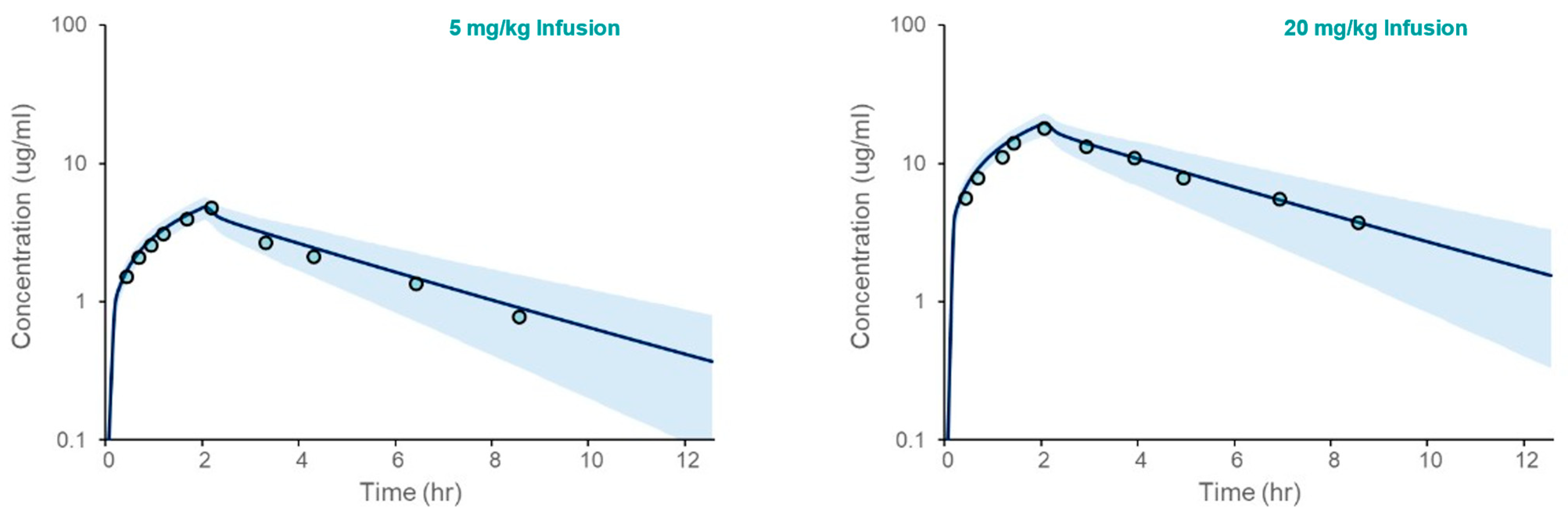
3.2. PBPK Model Verification
3.2.1. Simulations in Healthy Volunteers
3.2.2. Model Simulations in Pediatric Population
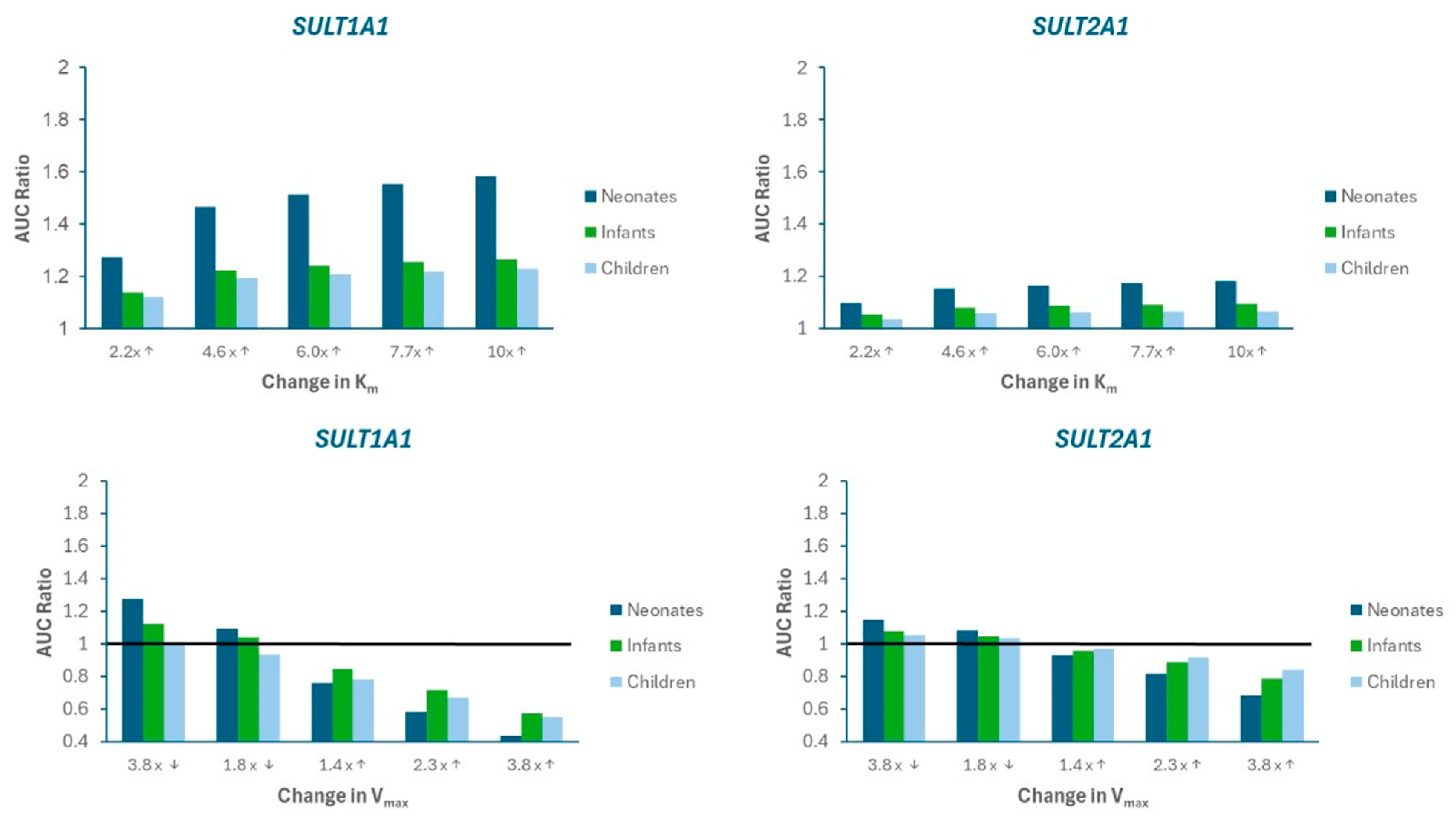
3.3. Model Sensitivity Analysis
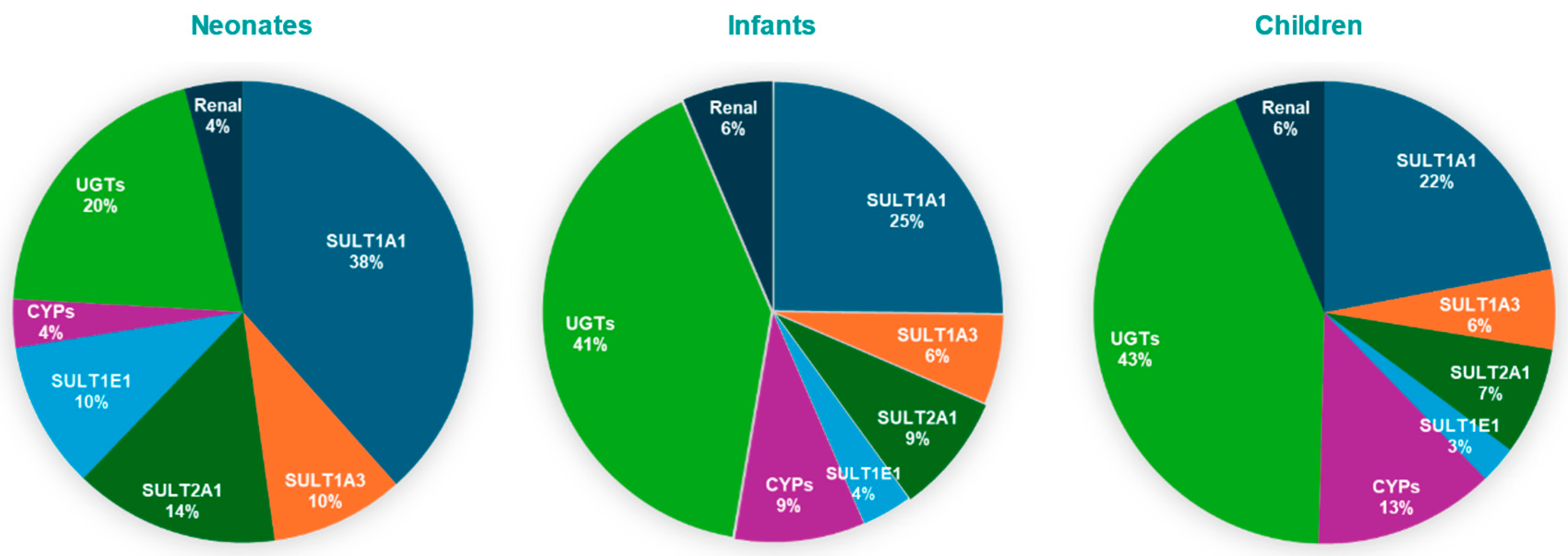
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rao, C.; Shenoy, V.; Udaykumar, P. Potential drug–drug interactions in the pediatric intensive care unit of a tertiary care hospital. J. Pharmacol. Pharmacother. 2019, 10, 63–68. [Google Scholar] [CrossRef]
- Hassanzad, M.; Arenas-Lopez, S.; Baniasadi, S. Potential drug–drug interactions among critically ill pediatric patients in a tertiary pulmonary center. J. Clin. Pharmacol. 2018, 58, 221–227. [Google Scholar] [CrossRef]
- Naji-Talakar, S.; Sharma, S.; Martin, L.A.; Barnhart, D.; Prasad, B. Potential implications of DMET ontogeny on the disposition of commonly prescribed drugs in neonatal and pediatric intensive care units. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 273–289. [Google Scholar] [CrossRef]
- Dai, D.; Feinstein, J.A.; Morrison, W.; Zuppa, A.F.; Feudtner, C. Epidemiology of polypharmacy and potential drug–drug interactions among pediatric patients in ICUs of U.S. children’s hospitals. Pediatr. Crit. Care Med. 2016, 17, e218–e228. [Google Scholar] [CrossRef] [PubMed]
- Golchin, N.; Johnson, H.; Bakaki, P.M.; Dawson, N.; Pestana Knight, E.M.; Meropol, S.B.; Liu, R.; Feinstein, J.A.; Bolen, S.D.; Kleinman, L.C.; et al. Outcome measures in pediatric polypharmacy research: A scoping review. Drugs Ther. Perspect. 2019, 35, 447–458. [Google Scholar] [CrossRef]
- James, M.O.; Ambadapadi, S. Interactions of cytosolic sulfotransferases with xenobiotics. Drug Metab. Rev. 2013, 45, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ladumor, M.K.; Bhatt, D.K.; Gaedigk, A.; Sharma, S.; Thakur, A.; Pearce, R.E.; Leeder, J.S.; Bolger, M.B.; Singh, S.; Prasad, B. Ontogeny of hepatic sulfotransferases and prediction of age-dependent fractional contribution of sulfation in acetaminophen metabolism. Drug Metab. Dispos. 2019, 47, 818–831. [Google Scholar] [CrossRef]
- Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 2008, 118, 250–267. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Liu, M.-Y.; Kurogi, K.; Sakakibara, Y.; Saeki, Y.; Suiko, M.; Liu, M.-C. Sulphation of acetaminophen by the human cytosolic sulfotransferases: A systematic analysis. J. Biochem. 2015, 158, 497–504. [Google Scholar] [CrossRef]
- Miller, R.P.; Roberts, R.J.; Fischer, L.J. Acetaminophen elimination kinetics in neonates, children, and adults. Clin. Pharmacol. Ther. 1976, 19, 284–294. [Google Scholar] [CrossRef]
- Ozbey, A.C.; Fowler, S.; Leys, K.; Annaert, P.; Umehara, K.; Parrott, N. Physiologically-Based pharmacokinetic modeling for drugs cleared by non-cytochrome p450 enzymes: State-of-the-art and future perspectives. Drug Metab. Dispos. 2024, 52, 44–55. [Google Scholar] [CrossRef]
- Stanley, E.L.; Hume, R.; Coughtrie, M.W.H. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol. Cell Endocrinol. 2005, 240, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Richard, K. Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J. Clin. Endocrinol. Metab. 2001, 86, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Dubaisi, S.; Barrett, K.G.; Fang, H.; Guzman-Lepe, J.; Soto-Gutierrez, A.; Kocarek, T.A.; Runge-Morris, M. Regulation of cytosolic sulfotransferases in models of human hepatocyte development. Drug Metab. Dispos. 2018, 46, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, P.; Barrett, J.; Lesko, L.; Schmidt, S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–9. [Google Scholar] [CrossRef]
- Cook, S.F.; Roberts, J.K.; Samiee-Zafarghandy, S.; Stockmann, C.; King, A.D.; Deutsch, N.; Williams, E.F.; Allegaert, K.; Wilkins, D.G.; Sherwin, C.M.T.; et al. Population pharmacokinetics of intravenous paracetamol (acetaminophen) in preterm and term neonates: Model development and external evaluation. Clin. Pharmacokinet. 2016, 55, 107–119. [Google Scholar] [CrossRef]
- Olafuyi, O.; Abbasi, M.Y.; Allegaert, K. Physiologically based pharmacokinetic modelling of acetaminophen in preterm neonates—The impact of metabolising enzyme ontogeny and reduced cardiac output. Biopharm. Drug Dispos. 2021, 42, 401–417. [Google Scholar] [CrossRef]
- Clements, J.; Critchley, J.; Prescott, L. The role of sulphate conjugation in the metabolism and disposition of oral and intravenous paracetamol in man. Br. J. Clin. Pharmacol. 1984, 18, 481–485. [Google Scholar] [CrossRef]
- Rohatgi, A. WebPlotDigitizer (Version 4.8) [Computer Software]. 2024. Available online: https://automeris.io (accessed on 6 June 2025).
- Zuppa, A.F.; Hammer, G.B.; Barrett, J.S.; Kenney, B.F.; Kassir, N.; Mouksassi, S.; Royal, M.A. Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or fever. J. Pediatr. Pharmacol. Ther. 2011, 16, 246–261. [Google Scholar] [CrossRef]
- Johnson, T.N.; Small, B.G.; Rowland Yeo, K. Increasing application of pediatric physiologically based pharmacokinetic models across academic and industry organizations. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 373–383. [Google Scholar] [CrossRef]
- Bhatt, D.K.; Mehrotra, A.; Gaedigk, A.; Chapa, R.; Basit, A.; Zhang, H.; Choudhari, P.; Boberg, M.; Pearce, R.E.; Gaedigk, R.; et al. Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin. Pharmacol. Ther. 2019, 105, 131–141. [Google Scholar] [CrossRef]
- Vander Schaaf, M.; Luth, K.; Townsend, D.M.; Chessman, K.H.; Mills, C.M.; Garner, S.S.; Peterson, Y.K. CYP3A4 drug metabolism considerations in pediatric pharmacotherapy. Med. Chem. Res. 2024, 33, 2221–2235. [Google Scholar] [CrossRef]
- Huisinga, W.; Solms, A.; Fronton, L.; Pilari, S. Modeling interindividual variability in physiologically based pharmacokinetics and its link to mechanistic covariate modeling. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Gaedigk, A.; Vrana, M.; Gaedigk, R.; Leeder, J.S.; Salphati, L.; Chu, X.; Xiao, G.; Hop, C.; Evers, R.; et al. Ontogeny of hepatic drug transporters as quantified by LC-MS/MS proteomics. Clin. Pharmacol. Ther. 2016, 100, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.; McNamara, P.J. Ontogeny of hepatic and renal systemic clearance pathways in infants: Part I. Clin. Pharmacokinet. 2002, 41, 959–998. [Google Scholar] [CrossRef]
- Riches, Z.; Stanley, E.L.; Bloomer, J.C.; Coughtrie, M.W.H. Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTS) in human tissues: The SULT “pie”. Drug Metab. Dispos. 2009, 37, 2255–2261. [Google Scholar] [CrossRef]
- Adjei, A.A.; Gaedigk, A.; Simon, S.D.; Weinshilboum, R.M.; Leeder, J.S. Interindividual variability in acetaminophen sulfation by human fetal liver: Implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 155–165. [Google Scholar] [CrossRef]
- Bansal, N.; Momin, S.; Bansal, R.; Gurram Venkata, S.K.R.; Ruser, L.; Yusuf, K. Pharmacokinetics of drugs: Newborn perspective. Pediatr. Med. 2024, 7, 19. [Google Scholar] [CrossRef]
- Van Der Marel, C.D.; Anderson, B.J.; Van Lingen, R.A.; Holford, N.H.G.; Pluim, M.A.L.; Jansman, F.G.A.; Van Den Anker, J.N.; Tibboel, D. Paracetamol and metabolite pharmacokinetics in infants. Eur. J. Clin. Pharmacol. 2003, 59, 243–251. [Google Scholar] [CrossRef]
- Flint, R.B.; Roofthooft, D.W.; Van Rongen, A.; Van Lingen, R.A.; Van Den Anker, J.N.; Van Dijk, M.; Allegaert, K.; Tibboel, D.; Knibbe, C.A.J.; Simons, S.H.P. Exposure to acetaminophen and all its metabolites upon 10, 15, and 20 mg/kg intravenous acetaminophen in very-preterm infants. Pediatr. Res. 2017, 82, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Knibbe, C.A.J.; Krekels, E.H.J.; Van Den Anker, J.N.; DeJongh, J.; Santen, G.W.E.; Van Dijk, M.; Simons, S.H.P.; Van Lingen, R.A.; Jacqz-Aigrain, E.M.; Danhof, M.; et al. Morphine glucuronidation in preterm neonates, infants and children younger than 3 years. Clin. Pharmacokinet. 2009, 48, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, G.M. Pharmacokinetics of cephalosporins in the neonate: A review. Clinics. 2011, 66, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.W.K.; Van Groen, B.D.; Burckart, G.J.; Zhang, L.; De Wildt, S.N.; Huang, S. Incorporating ontogeny in physiologically based pharmacokinetic modeling to improve pediatric drug development: What we know about developmental changes in membrane transporters. J. Clin. Pharmacol. 2019, 59, S56–S69. [Google Scholar] [CrossRef]
- Horace, A.; Ahmed, F. Polypharmacy in pediatric patients and opportunities for pharmacists involvement. Integr. Pharm. Res. Pract. 2015, 4, 113–126. [Google Scholar] [CrossRef]
- Mylan Pharmaceuticals Inc. Meclofenamate Sodium [Package Insert]; Mylan Pharmaceuticals Inc.: Morgantown, WV, USA, 2024. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter a | Value |
|---|---|
| Physicochemical Properties | |
| Molecular weight | 151.2 g/mol |
| LogP | 0.51 |
| pKa | 9.46 |
| Fraction unbound in plasma | 0.82 (Bound to albumin) |
| Blood/plasma ratio | 1 |
| Absorption | |
| Absorption Model | ADAM |
| Peff | 12 |
| ka | 5.24 |
| fa | 0.99 |
| Dissolution | |
| Solubility (mg/mL) at pH 8.94 | 13.65 |
| Distribution | |
| Distribution Model | Minimal PBPK model |
| Vss (L/kg) b | 0.8 |
| Kp scalar b | 1.63 |
| Metabolism/Elimination | |
| Clearance Type | Enzyme Kinetics |
| UGT 1A1 | Vmax = 6654; Km = 5500 |
| UGT1A9 | Vmax = 11,130; Km = 9200 |
| UGT2B15 | Vmax = 37,101; Km = 23,000 |
| SULT 1A1 | Vmax = 1549; Km = 2400 |
| SULT 1A3 | Vmax = 231.0; Km = 1500 |
| SULT1E1 | Vmax = 167.2; Km = 1900 |
| SULT2A1 | Vmax = 814.1; Km = 3700 |
| CYP1A2 | Vmax = 34.5; Km = 220 |
| CYP2C9 | Vmax = 9.86; Km = 660 |
| CYP2C19 | Vmax = 29.87 Km = 2000 |
| CYP2D6 | Vmax = 6.57; Km = 440 |
| CTP2E1 | Vmax = 90.06; Km = 4020 |
| CYP3A4 | Vmax = 62.13; Km = 130 |
| ClIV (L/h) | 19.7 |
| ClR (L/h) | 1.12 |
| Active hepatic scalar | 1.5 |
| Enzyme | Ontogeny Equation * | |
|---|---|---|
| SULT1A1 | [age < 1.8 years] | |
| [1.83 ≤ age ≤ 12.6 years] | ||
| [age > 12.6 years] | ||
| SULT1A3 | [age < 2 years] | |
| [2 ≤ age ≤ 8.6 years] | ||
| [age > 8.6 years] | ||
| SULT2A1 | [age < 1.2 years] | |
| [1.2 ≤ age ≤ 13.2 years] | ||
| [age > 13.2 years] | ||
| SULT1B1 | [age < 2.6 years] | |
| [2.6 ≤ age ≤ 8.1 years] | ||
| [age > 8.1 years] | ||
| Dose | Parameter | PBPK Model-Predicted | Observed * | Fold Error |
|---|---|---|---|---|
| 5 mg/kg Infusion | Cmax (μg/mL) | 4.95 ± 0.57 | 4.74 | 1.04 |
| AUC0-∞ (µg-hr/mL) | 22.9 ± 6.15 | 18.4 ± 1.65 | 1.24 | |
| 20 mg/kg Infusion | Cmax (μg/mL) | 20.0 ± 2.27 | 17.8 | 1.12 |
| AUC0-∞ (µg-hr/mL) | 93.5 ± 25.3 | 82.5 ± 10.0 | 1.13 |
| Population (Dosing Regimen) | Parameter * | PBPK Model Predicted | Observed | Fold Error |
|---|---|---|---|---|
| Children (12.5 mg/kg Q 6h) | Cmax (μg/mL) | 28.6 (23.9–40.2) | 24.3 (3.8–35.1) | 1.18 |
| AUC0-τ (µg-hr/mL) | 35.7 (20.7–76.4) | 37.8 (11.3–52.3) | 0.94 | |
| Infants (12.5 mg/kg Q 4h) | Cmax (μg/mL) | 22.8 (18.4–30.2) | 21.9 (4.2–25.3) | 1.04 |
| AUC0-τ (µg-hr/mL) | 39.1 (22.7–69.3) | 43.3 (9.2–79.2) | 0.90 | |
| Neonates (12.5 mg/kg Q 4h) | Cmax (μg/mL) | 21.8 (17.7–28.5) | 19.9 (19.3–20.5) | 1.10 |
| AUC0-τ (µg-hr/mL) | 53.6 (26.7–92.1) | 65.6 (55.8–75.4) | 0.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Taft, D.R. PBPK Modeling of Acetaminophen in Pediatric Populations: Incorporation of SULT Enzyme Ontogeny to Predict Age-Dependent Metabolism and Systemic Exposure. Life 2025, 15, 1099. https://doi.org/10.3390/life15071099
Sharma S, Taft DR. PBPK Modeling of Acetaminophen in Pediatric Populations: Incorporation of SULT Enzyme Ontogeny to Predict Age-Dependent Metabolism and Systemic Exposure. Life. 2025; 15(7):1099. https://doi.org/10.3390/life15071099
Chicago/Turabian StyleSharma, Sonia, and David R. Taft. 2025. "PBPK Modeling of Acetaminophen in Pediatric Populations: Incorporation of SULT Enzyme Ontogeny to Predict Age-Dependent Metabolism and Systemic Exposure" Life 15, no. 7: 1099. https://doi.org/10.3390/life15071099
APA StyleSharma, S., & Taft, D. R. (2025). PBPK Modeling of Acetaminophen in Pediatric Populations: Incorporation of SULT Enzyme Ontogeny to Predict Age-Dependent Metabolism and Systemic Exposure. Life, 15(7), 1099. https://doi.org/10.3390/life15071099