

Galectin-3 Release in the Bone Marrow Microenvironment Promotes Drug Resistance and Relapse in Acute Myeloid Leukemia


Abstract

1. Introduction
2. Acute Myeloid Leukemia
2.1. High BM Gal-3 Expression and Plasma Gal-3 Levels Are Associated with Poor Prognosis
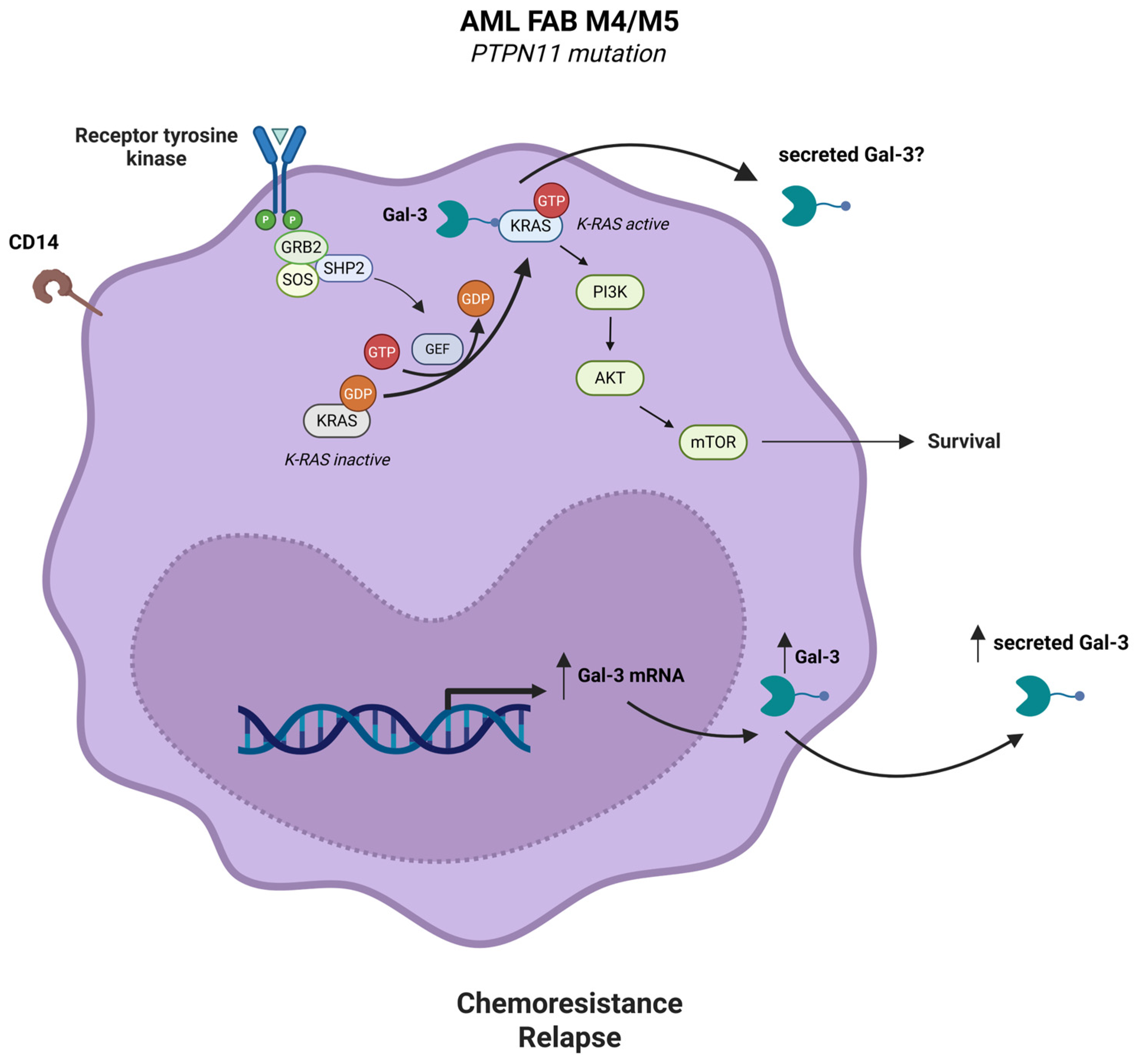
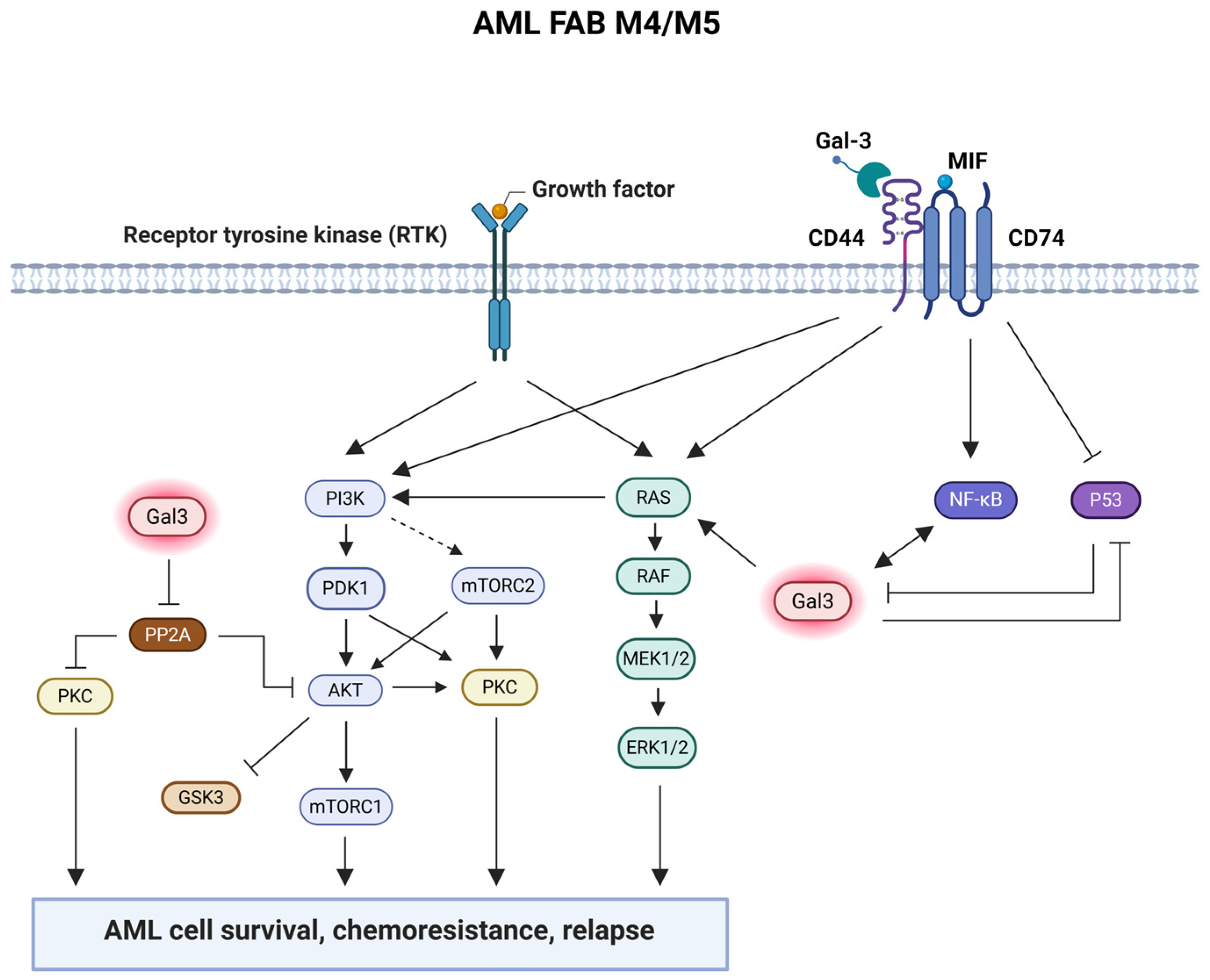
2.2. Gal-3 Is Highly Expressed by AML Cells and Synergizes with CD74/CD44 Signaling Pathway to Regulate AML Cell Survival
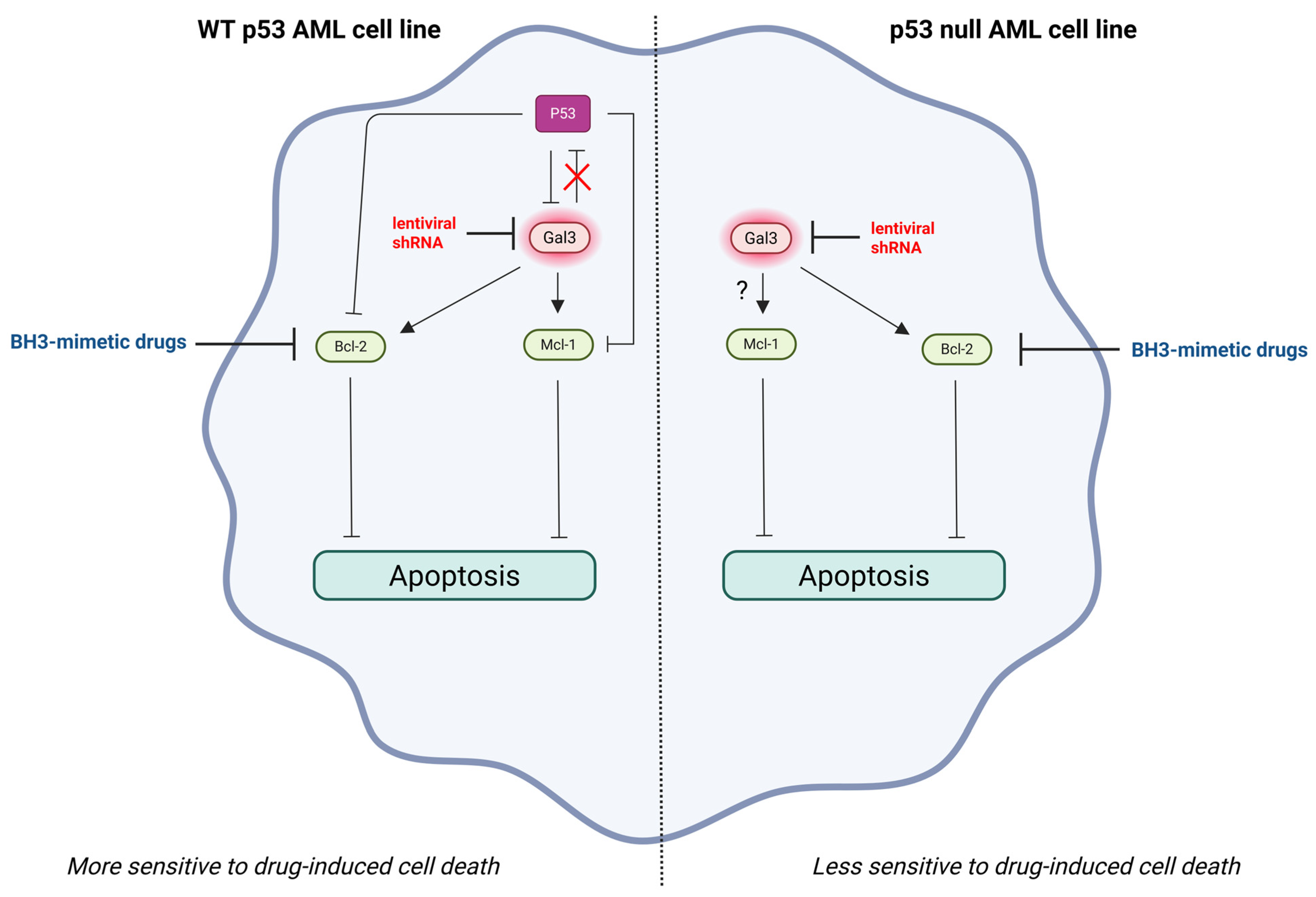
2.3. Gal-3 Expression in AML Cells Promotes Chemotherapeutic Resistance by Stimulating AML Cell Survival
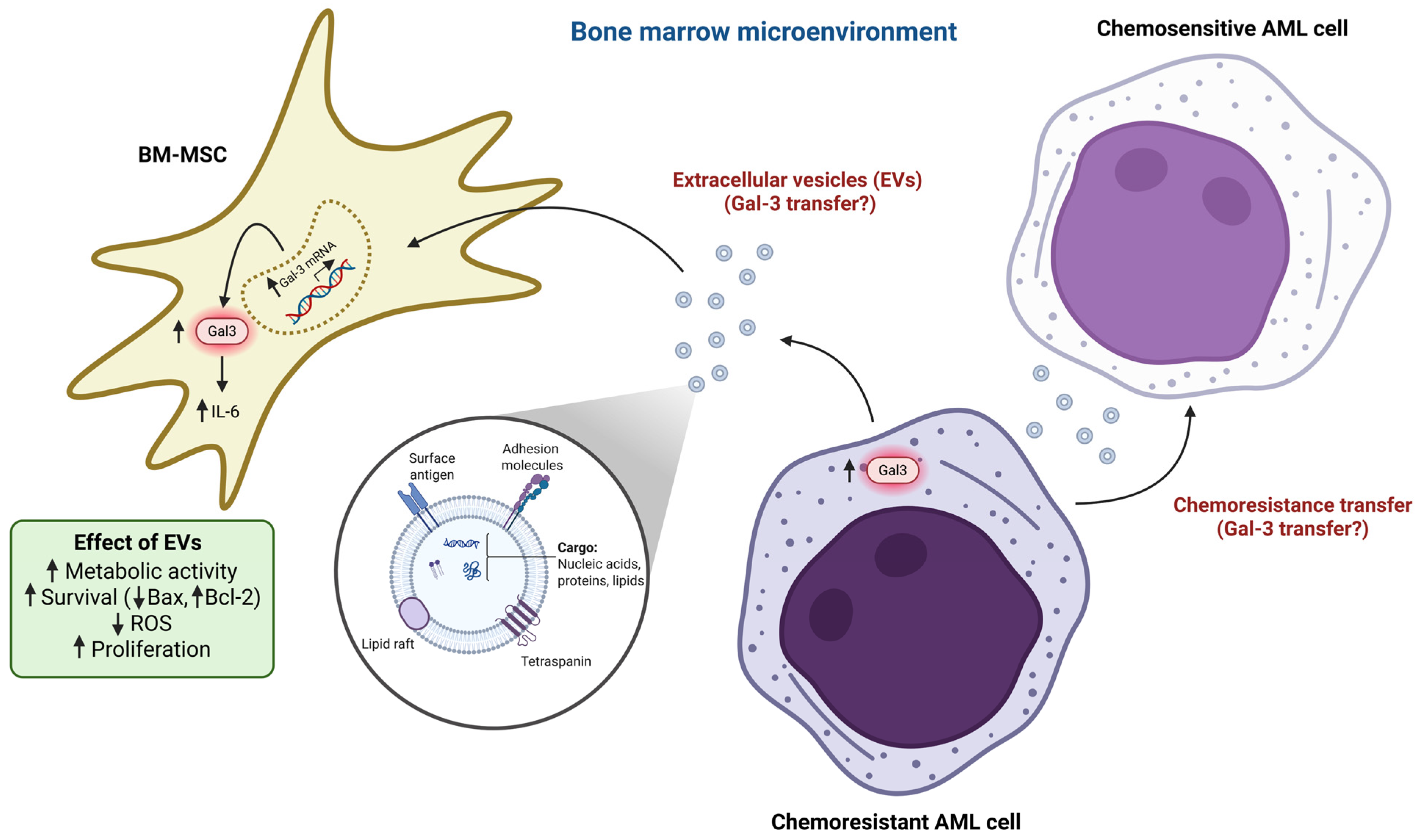
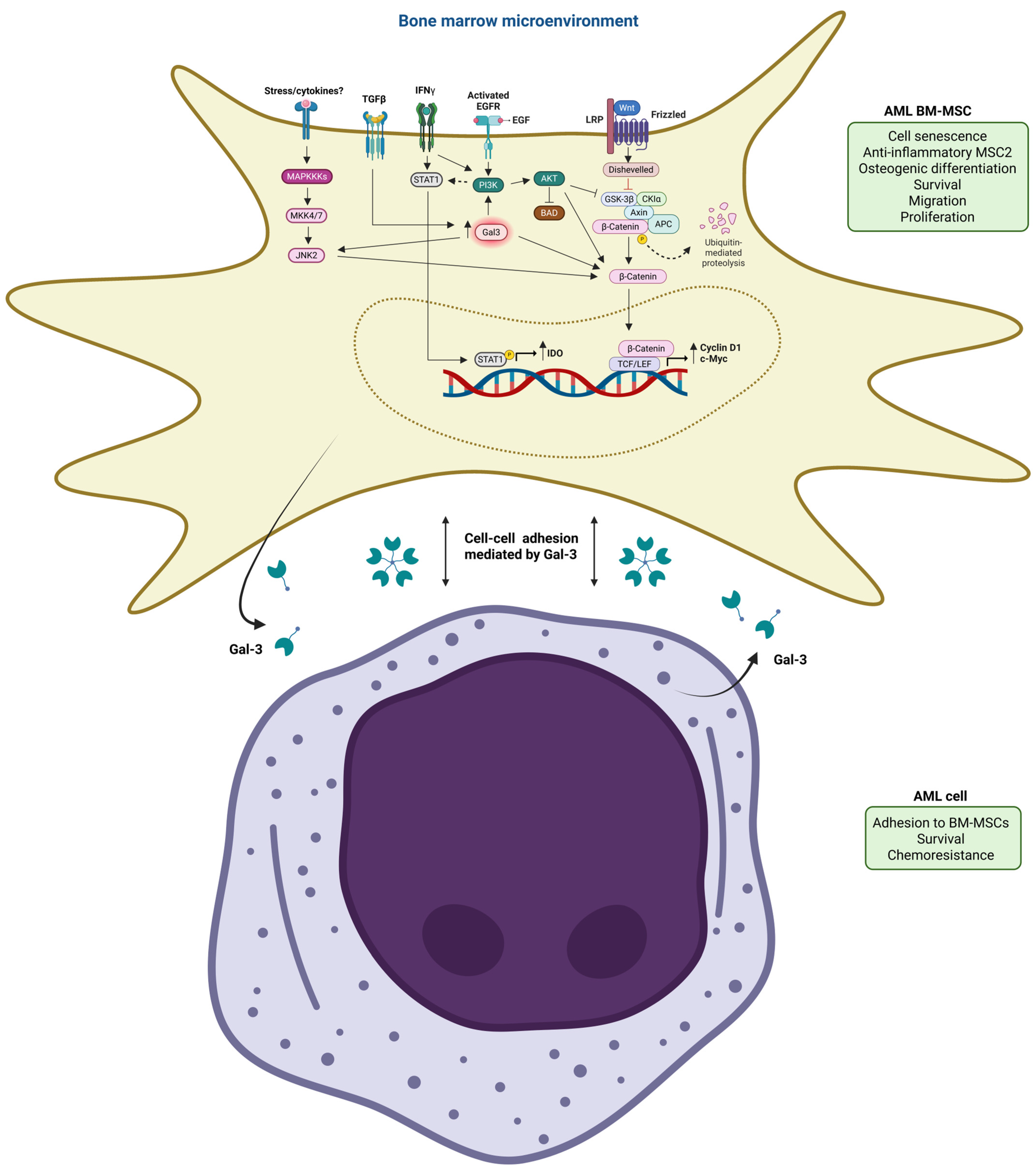
2.4. BM-MSCs Induce Drug Resistance of AML Cells by Gal-3 Upregulation in AML Cells
2.5. AML Cell-Derived Extracellular Vesicles Upregulate Gal-3 Expression in BM-MSCs
2.6. BM-MSCs Derived from AML Patients Highly Express Gal-3 Protein During Relapse
2.7. BM-MSC-Derived Gal-3 Promotes AML Cell Adhesion and Survival
3. Therapeutic Targeting of Gal-3: Insights Gained from Other Diseases
4. Future Directions
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azizidoost, S.; Babashah, S.; Rahim, F.; Shahjahani, M.; Saki, N. Bone Marrow Neoplastic Niche in Leukemia. Hematology 2014, 19, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and Leukemic Stem Cell Niches: Insights and Therapeutic Opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef]
- Konopleva, M.; Tabe, Y.; Zeng, Z.; Andreeff, M. Therapeutic Targeting of Microenvironmental Interactions in Leukemia: Mechanisms and Approaches. Drug Resist. Updates 2009, 12, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Advances in Understanding the Leukaemia Microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-Mediated Drug Resistance: A Major Contributor to Minimal Residual Disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Nair, R.R.; Tolentino, J.; Hazlehurst, L.A. The Bone Marrow Microenvironment as a Sanctuary for Minimal Residual Disease in CML. Biochem. Pharmacol. 2010, 80, 602–612. [Google Scholar] [CrossRef]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castaño, J.; Gómez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Hou, H.A.; Lee, M.C.; Liu, C.Y.; Jhuang, J.Y.; Lai, Y.J.; Lin, C.W.; Chen, H.Y.; Liu, F.T.; Chou, W.C.; et al. Higher Bone Marrow LGALS3 Expression Is an Independent Unfavorable Prognostic Factor for Overall Survival in Patients with Acute Myeloid Leukemia. Blood 2013, 121, 3172–3180. [Google Scholar] [CrossRef]
- Fei, F.; Abdel-Azim, H.; Lim, M.; Arutyunyan, A.; von Itzstein, M.; Groffen, J.; Heisterkamp, N. Galectin-3 in Pre-B Acute Lymphoblastic Leukemia. Leukemia 2013, 27, 2385–2388. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-Cell Precursor Acute Lymphoblastic Leukemia and Stromal Cells Communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef]
- Yamamoto-Sugitani, M.; Kuroda, J.; Ashihara, E.; Nagoshi, H.; Kobayashi, T.; Matsumoto, Y.; Sasaki, N.; Shimura, Y.; Kiyota, M.; Nakayama, R.; et al. Galectin-3 (Gal-3) Induced by Leukemia Microenvironment Promotes Drug Resistance and Bone Marrow Lodgment in Chronic Myelogenous Leukemia. Proc. Natl. Acad. Sci. USA 2011, 108, 17468–17473. [Google Scholar] [CrossRef] [PubMed]
- Asgarian-Omran, H.; Forghani, P.; Hojjat-Farsangi, M.; Roohi, A.; Sharifian, R.A.; Razavi, S.M.; Jeddi-Tehrani, M.; Rabbani, H.; Shokri, F. Expression Profile of Galectin-1 and Galectin-3 Molecules in Different Subtypes of Chronic Lymphocytic Leukemia. Cancer Investig. 2010, 28, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Toscano, M.A.; Jackson, S.S.; Vasta, G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007, 17, 513–520. [Google Scholar] [CrossRef]
- Ko, F.C.F.; Yan, S.; Lee, K.W.; Lam, S.K.; Ho, J.C.M. Chimera and Tandem-Repeat Type Galectins: The New Targets for Cancer Immunotherapy. Biomolecules 2023, 13, 902. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Galectin 3 as a Guardian of the Tumor Microenvironment. Biochim. Biophys. Acta 2016, 1863, 427–437. [Google Scholar] [CrossRef]
- Liu, F.-T.; Stowell, S.R. The Role of Galectins in Immunity and Infection. Nat. Rev. Immunol. 2023, 23, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Balan, V.; Raz, A. Regulation of Tumor Progression by Extracellular Galectin-3. Cancer Microenviron. 2008, 1, 43–51. [Google Scholar] [CrossRef]
- Ahmed, R.; Anam, K.; Ahmed, H. Development of Galectin-3 Targeting Drugs for Therapeutic Applications in Various Diseases. Int. J. Mol. Sci. 2023, 24, 8116. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Hogan, V.; Raz, A. Galectin-3 and Cancer Stemness. Glycobiology 2018, 28, 172–181. [Google Scholar] [CrossRef]
- Sundblad, V.; Croci, D.O.; Rabinovich, G.A. Regulated Expression of Galectin-3, a Multifunctional Glycan-Binding Protein, in Haematopoietic and Non-Haematopoietic Tissues. Histol. Histopathol. 2011, 26, 247–265. [Google Scholar] [CrossRef]
- Liu, F.T.; Hsu, D.K.; Zuberi, R.I.; Kuwabara, I.; Chi, E.Y.; Henderson, W.R. Expression and Function of Galectin-3, a Beta-Galactoside-Binding Lectin, in Human Monocytes and Macrophages. Am. J. Pathol. 1995, 147, 1016–1028. [Google Scholar]
- Dietz, A.B.; Bulur, P.A.; Knutson, G.J.; Matasić, R.; Vuk-Pavlović, S. Maturation of Human Monocyte-Derived Dendritic Cells Studied by Microarray Hybridization. Biochem. Biophys. Res. Commun. 2000, 275, 731–738. [Google Scholar] [CrossRef]
- Truong, M.J.; Gruart, V.; Kusnierz, J.P.; Papin, J.P.; Loiseau, S.; Capron, A.; Capron, M. Human Neutrophils Express Immunoglobulin E (IgE)-Binding Proteins (Mac-2/Epsilon BP) of the S-Type Lectin Family: Role in IgE-Dependent Activation. J. Exp. Med. 1993, 177, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Truong, M.; Gruart, V.; Liu, F.; Prin, L.; Capron, A.; Capron, M. IgE-binding Molecules (Mac-2/ΕBP) Expressed by Human Eosinophils. Implication in IgE-dependent Eosinophil Cytotoxicity. Eur. J. Immunol. 1993, 23, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.S.; Krishnaswamy, P.; Irani, A.A.; Kepley, C.L.; Liu, F.; Schwartz, L.B. Immunoelectron Microscopic Localization of Galectin-3, an IgE Binding Protein, in Human Mast Cells and Basophils. Anat. Rec. 1995, 242, 211–219. [Google Scholar] [CrossRef]
- Brittoli, A.; Fallarini, S.; Zhang, H.; Pieters, R.J.; Lombardi, G. “In Vitro” Studies on Galectin-3 in Human Natural Killer Cells. Immunol. Lett. 2018, 194, 4–12. [Google Scholar] [CrossRef]
- Hsu, D.K.; Hammes, S.R.; Kuwabara, I.; Greene, W.C.; Liu, F.T. Human T Lymphotropic Virus-I Infection of Human T Lymphocytes Induces Expression of the Beta-Galactoside-Binding Lectin, Galectin-3. Am. J. Pathol. 1996, 148, 1661–1670. [Google Scholar] [PubMed]
- Fogel, S.; Guittaut, M.; Legrand, A.; Monsigny, M.; Hebert, E. The Tat Protein of HIV-1 Induces Galectin-3 Expression. Glycobiology 1999, 9, 383–387. [Google Scholar] [CrossRef]
- Menon, R.P.; Hughes, R.C. Determinants in the N-Terminal Domains of Galectin-3 for Secretion by a Novel Pathway Circumventing the Endoplasmic Reticulum-Golgi Complex. Eur. J. Biochem. 1999, 264, 569–576. [Google Scholar] [CrossRef]
- Hughes, R.C. Secretion of the Galectin Family of Mammalian Carbohydrate-Binding Proteins. Biochim. Biophys. Acta 1999, 1473, 172–185. [Google Scholar] [CrossRef]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An Open-Ended Story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Fortuna-Costa, A.; Gomes, A.M.; Kozlowski, E.O.; Stelling, M.P.; Pavão, M.S.G. Extracellular Galectin-3 in Tumor Progression and Metastasis. Front. Oncol. 2014, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Finley, R.L.; Raz, A.; Kim, H.-R.C. Galectin-3 Translocates to the Perinuclear Membranes and Inhibits Cytochrome c Release from the Mitochondria. A Role for Synexin in Galectin-3 Translocation. J. Biol. Chem. 2002, 277, 15819–15827. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Hogan, V.; Inohara, H.; Raz, A. Importin-Mediated Nuclear Translocation of Galectin-3. J. Biol. Chem. 2006, 281, 39649–39659. [Google Scholar] [CrossRef]
- Yoshii, T.; Fukumori, T.; Honjo, Y.; Inohara, H.; Kim, H.-R.C.; Raz, A. Galectin-3 Phosphorylation Is Required for Its Anti-Apoptotic Function and Cell Cycle Arrest. J. Biol. Chem. 2002, 277, 6852–6857. [Google Scholar] [CrossRef]
- Takenaka, Y.; Fukumori, T.; Yoshii, T.; Oka, N.; Inohara, H.; Kim, H.-R.C.; Bresalier, R.S.; Raz, A. Nuclear Export of Phosphorylated Galectin-3 Regulates Its Antiapoptotic Activity in Response to Chemotherapeutic Drugs. Mol. Cell Biol. 2004, 24, 4395–4406. [Google Scholar] [CrossRef]
- Balan, V.; Nangia-Makker, P.; Jung, Y.S.; Wang, Y.; Raz, A. Galectin-3: A Novel Substrate for c-Abl Kinase. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 1198–1205. [Google Scholar] [CrossRef]
- Balan, V.; Nangia-Makker, P.; Kho, D.H.; Wang, Y.; Raz, A. Tyrosine-Phosphorylated Galectin-3 Protein Is Resistant to Prostate-Specific Antigen (PSA) Cleavage. J. Biol. Chem. 2012, 287, 5192–5198. [Google Scholar] [CrossRef]
- Nakahara, S.; Oka, N.; Raz, A. On the Role of Galectin-3 in Cancer Apoptosis. Apoptosis 2005, 10, 267–275. [Google Scholar] [CrossRef]
- Farhad, M.; Rolig, A.S.; Redmond, W.L. The Role of Galectin-3 in Modulating Tumor Growth and Immunosuppression within the Tumor Microenvironment. Oncoimmunology 2018, 7, e1434467. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a Novel Biomarker for Disease Diagnosis and a Target for Therapy (Review). Int. J. Mol. Med. 2017, 41, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Haudek, K.C.; Spronk, K.J.; Voss, P.G.; Patterson, R.J.; Wang, J.L.; Arnoys, E.J. Dynamics of Galectin-3 in the Nucleus and Cytoplasm. Biochim. Biophys. Acta (BBA)-General. Subj. 2010, 1800, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Newlaczyl, A.U.; Yu, L.-G. Galectin-3—A Jack-of-All-Trades in Cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef]
- Gao, N.; Wang, X.-X.; Sun, J.-R.; Yu, W.-Z.; Li, X.-Z. Clinical Impact of Galectin-3 in Newly Diagnosed t (15;17)(Q22;Q21)/PML-RARa Acute Promyelocytic Leukemia Treated with All-Trans Retinoic Acid and Arsenic Trioxide-Based Regimens. Ann. Hematol. 2017, 96, 711–718. [Google Scholar] [CrossRef]
- Gao, N.; Yu, W.-Z.; Guo, N.-J.; Wang, X.-X.; Sun, J.-R. Clinical Significance of Galectin-3 in Patients with Adult Acute Myeloid Leukemia: A Retrospective Cohort Study with Long-Term Follow-up and Formulation of Risk Scoring System. Leuk. Lymphoma 2017, 58, 1394–1402. [Google Scholar] [CrossRef]
- Wang, J.; Gao, N.; Wang, X.; Yu, W.; Li, A. Prognostic Factors in Acute Myeloid Leukemia with t(8;21)/AML1-ETO: Strategies to Define High-Risk Patients. Indian J. Hematol. Blood Transfus. 2022, 38, 631–637. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Shannon, K.M.; Loh, M.L. SHP-2 and Myeloid Malignancies. Curr. Opin. Hematol. 2004, 11, 44–50. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic Mutations in PTPN11 in Juvenile Myelomonocytic Leukemia, Myelodysplastic Syndromes and Acute Myeloid Leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.-A.; Chou, W.-C.; Lin, L.-I.; Chen, C.-Y.; Tang, J.-L.; Tseng, M.-H.; Huang, C.-F.; Chiou, R.-J.; Lee, F.-Y.; Liu, M.-C.; et al. Characterization of Acute Myeloid Leukemia with PTPN11 Mutation: The Mutation Is Closely Associated with NPM1 Mutation but Inversely Related to FLT3/ITD. Leukemia 2008, 22, 1075–1078. [Google Scholar] [CrossRef]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 Augments K-Ras Activation and Triggers a Ras Signal That Attenuates ERK but Not Phosphoinositide 3-Kinase Activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef]
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.J.; van Putten, W.L.J.; Valk, P.J.M.; Delwel, R. Double CEBPA Mutations, but Not Single CEBPA Mutations, Define a Subgroup of Acute Myeloid Leukemia with a Distinctive Gene Expression Profile That Is Uniquely Associated with a Favorable Outcome. Blood 2009, 113, 3088–3091. [Google Scholar] [CrossRef]
- Taskesen, E.; Bullinger, L.; Corbacioglu, A.; Sanders, M.A.; Erpelinck, C.A.J.; Wouters, B.J.; van der Poel-van de Luytgaarde, S.C.; Damm, F.; Krauter, J.; Ganser, A.; et al. Prognostic Impact, Concurrent Genetic Mutations, and Gene Expression Features of AML with CEBPA Mutations in a Cohort of 1182 Cytogenetically Normal AML Patients: Further Evidence for CEBPA Double Mutant AML as a Distinctive Disease Entity. Blood 2011, 117, 2469–2475. [Google Scholar] [CrossRef]
- Yuan, J.; He, R.; Alkhateeb, H.B. Sporadic and Familial Acute Myeloid Leukemia with CEBPA Mutations. Curr. Hematol. Malig. Rep. 2023, 18, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Lo Coco, F.; Martín, G.; Avvisati, G.; Rayón, C.; Barbui, T.; Díaz-Mediavilla, J.; Fioritoni, G.; González, J.D.; Liso, V.; et al. Definition of Relapse Risk and Role of Nonanthracycline Drugs for Consolidation in Patients with Acute Promyelocytic Leukemia: A Joint Study of the PETHEMA and GIMEMA Cooperative Groups. Blood 2000, 96, 1247–1253. [Google Scholar] [PubMed]
- Avvisati, G.; Lo-Coco, F.; Paoloni, F.P.; Petti, M.C.; Diverio, D.; Vignetti, M.; Latagliata, R.; Specchia, G.; Baccarani, M.; Di Bona, E.; et al. AIDA 0493 Protocol for Newly Diagnosed Acute Promyelocytic Leukemia: Very Long-Term Results and Role of Maintenance. Blood 2011, 117, 4716–4725. [Google Scholar] [CrossRef]
- Lehmann, S.; Ravn, A.; Carlsson, L.; Antunovic, P.; Deneberg, S.; Möllgård, L.; Rangert Derolf, Å.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; et al. Continuing High Early Death Rate in Acute Promyelocytic Leukemia: A Population-Based Report from the Swedish Adult Acute Leukemia Registry. Leukemia 2011, 25, 1128–1134. [Google Scholar] [CrossRef]
- Chendamarai, E.; Ganesan, S.; Alex, A.A.; Kamath, V.; Nair, S.C.; Nellickal, A.J.; Janet, N.B.; Srivastava, V.; Lakshmi, K.M.; Viswabandya, A.; et al. Comparison of Newly Diagnosed and Relapsed Patients with Acute Promyelocytic Leukemia Treated with Arsenic Trioxide: Insight into Mechanisms of Resistance. PLoS ONE 2015, 10, e0121912. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective Evaluation of Gene Mutations and Minimal Residual Disease in Patients with Core Binding Factor Acute Myeloid Leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of Cytogenetic Classification in Acute Myeloid Leukemia: Determination of Prognostic Significance of Rare Recurring Chromosomal Abnormalities among 5876 Younger Adult Patients Treated in the United Kingdom Medical Research Council Trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Schaich, M.; Koch, R.; Soucek, S.; Repp, R.; Ehninger, G.; Illmer, T. A Sensitive Model for Prediction of Relapse in Adult Acute Myeloid Leukaemia with t(8;21) Using White Blood Cell Count, CD56 and MDR1 Gene Expression at Diagnosis. Br. J. Haematol. 2004, 125, 477–479. [Google Scholar] [CrossRef]
- Wang, L.; Guo, X.-L. Molecular Regulation of Galectin-3 Expression and Therapeutic Implication in Cancer Progression. Biomed. Pharmacother. 2016, 78, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Sasca, D.; Szybinski, J.; Schüler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.-M.; Gebhardt, W.H.; et al. NCAM1 (CD56) Promotes Leukemogenesis and Confers Drug Resistance in AML. Blood 2019, 133, 2305–2319. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Balan, V.; Tai, G.; Raz, A. Galectin-3 Induces Cell Migration via a Calcium-Sensitive MAPK/ERK1/2 Pathway. Oncotarget 2014, 5, 2077–2084. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Hu, C.W.; Qiu, Y.; Ruvolo, V.R.; Go, R.L.; Hubner, S.E.; Coombes, K.R.; Andreeff, M.; Qutub, A.A.; Kornblau, S.M. LGALS3 Is Connected to CD74 in a Previously Unknown Protein Network That Is Associated with Poor Survival in Patients with AML. EBioMedicine 2019, 44, 126–137. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef]
- De Stefano, A.; Marvi, M.V.; Fazio, A.; McCubrey, J.A.; Suh, P.-G.; Ratti, S.; Ramazzotti, G.; Manzoli, L.; Cocco, L.; Follo, M.Y. Advances in MDS/AML and Inositide Signalling. Adv. Biol. Regul. 2023, 87, 100955. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.B.d.C.; Costa, R.G.A.; Silva, S.L.R.; Dias, I.R.S.B.; Dias, R.B.; Bezerra, D.P. Cell Signaling Pathways as Molecular Targets to Eliminate AML Stem Cells. Crit. Rev. Oncol. Hematol. 2021, 160, 103277. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; McQueen, T.; Chism, D.; Milella, M.; Estey, E.; Kaldjian, E.; Sebolt-Leopold, J.; Konopleva, M.; Andreeff, M. Quantitative Single Cell Determination of ERK Phosphorylation and Regulation in Relapsed and Refractory Primary Acute Myeloid Leukemia. Leukemia 2005, 19, 1543–1549. [Google Scholar] [CrossRef]
- Ikeda, D.; Chi, S.; Uchiyama, S.; Nakamura, H.; Guo, Y.-M.; Yamauchi, N.; Yuda, J.; Minami, Y. Molecular Classification and Overcoming Therapy Resistance for Acute Myeloid Leukemia with Adverse Genetic Factors. Int. J. Mol. Sci. 2022, 23, 5950. [Google Scholar] [CrossRef]
- Hayun, M.; Zaatra, M.; Itzkovich, C.; Sahar, D.; Rosenberg, D.; Filatova, M.; Ringelstein-Harlev, S.; Baris, H.; Moustafa-Hawash, N.; Louria-Hayon, I.; et al. ERK Activity in Immature Leukemic Cells Drives Clonal Selection during Induction Therapy for Acute Myeloid Leukemia. Sci. Rep. 2020, 10, 8349. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous Activation of Multiple Signal Transduction Pathways Confers Poor Prognosis in Acute Myelogenous Leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Qiu, Y.; Coombes, K.R.; Zhang, N.; Neeley, E.S.; Ruvolo, V.R.; Hail, N.; Borthakur, G.; Konopleva, M.; Andreeff, M.; et al. Phosphorylation of GSK3α/β Correlates with Activation of AKT and Is Prognostic for Poor Overall Survival in Acute Myeloid Leukemia Patients. BBA Clin. 2015, 4, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Pal, D. Two Faces of Protein Kinase Cδ: The Contrasting Roles of PKCδ in Cell Survival and Cell Death. Sci. World J. 2010, 10, 2272–2284. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Zhou, L.; Watt, J.C.; Ruvolo, V.R.; Burks, J.K.; Jiffar, T.; Kornblau, S.; Konopleva, M.; Andreeff, M. Targeting PKC-Mediated Signal Transduction Pathways Using Enzastaurin to Promote Apoptosis in Acute Myeloid Leukemia-Derived Cell Lines and Blast Cells. J. Cell Biochem. 2011, 112, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, V.R.; Karanjeet, K.B.; Schuster, T.F.; Brown, R.; Deng, Y.; Hinchcliffe, E.; Ruvolo, P.P. Role for PKC in Fenretinide-Mediated Apoptosis in Lymphoid Leukemia Cells. J. Signal Transduct. 2010, 2010, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kinehara, M.; Kawamura, S.; Tateyama, D.; Suga, M.; Matsumura, H.; Mimura, S.; Hirayama, N.; Hirata, M.; Uchio-Yamada, K.; Kohara, A.; et al. Protein Kinase C Regulates Human Pluripotent Stem Cell Self-Renewal. PLoS ONE 2013, 8, e54122. [Google Scholar] [CrossRef]
- Voisset, E.; Brenet, F.; Lopez, S.; de Sepulveda, P. SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis. Cancers 2020, 12, 1996. [Google Scholar] [CrossRef]
- El-Sisi, M.G.; Radwan, S.M.; Saeed, A.M.; El-Mesallamy, H.O. Serum Levels of FAK and Some of Its Effectors in Adult AML: Correlation with Prognostic Factors and Survival. Mol. Cell Biochem. 2021, 476, 1949–1963. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Qui, Y.H.; Coombes, K.R.; Zhang, N.; Ruvolo, V.R.; Borthakur, G.; Konopleva, M.; Andreeff, M.; Kornblau, S.M. Low Expression of PP2A Regulatory Subunit B55α Is Associated with T308 Phosphorylation of AKT and Shorter Complete Remission Duration in Acute Myeloid Leukemia Patients. Leukemia 2011, 25, 1711–1717. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of Protein Kinase Cascades by Protein Phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Seguin, L.; Camargo, M.F.; Wettersten, H.I.; Kato, S.; Desgrosellier, J.S.; von Schalscha, T.; Elliott, K.C.; Cosset, E.; Lesperance, J.; Weis, S.M.; et al. Galectin-3, a Druggable Vulnerability for KRAS-Addicted Cancers. Cancer Discov. 2017, 7, 1464–1479. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Ruvolo, V.R.; Benton, C.B.; AlRawi, A.; Burks, J.K.; Schober, W.; Rolke, J.; Tidmarsh, G.; Hail, N.; Eric Davis, R.; et al. Combination of Galectin Inhibitor GCS-100 and BH3 Mimetics Eliminates Both P53 Wild Type and P53 Null AML Cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving Complexity of MIF Signaling. Cell Signal 2019, 57, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Penticuff, J.C.; Woolbright, B.L.; Sielecki, T.M.; Weir, S.J.; Taylor, J.A. MIF Family Proteins in Genitourinary Cancer: Tumorigenic Roles and Therapeutic Potential. Nat. Rev. Urol. 2019, 16, 318–328. [Google Scholar] [CrossRef]
- Schröder, B. The Multifaceted Roles of the Invariant Chain CD74—More than Just a Chaperone. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 1269–1281. [Google Scholar] [CrossRef]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF Signal Transduction Initiated by Binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef]
- Lue, H.; Kapurniotu, A.; Fingerle-Rowson, G.; Roger, T.; Leng, L.; Thiele, M.; Calandra, T.; Bucala, R.; Bernhagen, J. Rapid and Transient Activation of the ERK MAPK Signalling Pathway by Macrophage Migration Inhibitory Factor (MIF) and Dependence on JAB1/CSN5 and Src Kinase Activity. Cell Signal 2006, 18, 688–703. [Google Scholar] [CrossRef]
- Lue, H.; Thiele, M.; Franz, J.; Dahl, E.; Speckgens, S.; Leng, L.; Fingerle-Rowson, G.; Bucala, R.; Lüscher, B.; Bernhagen, J. Macrophage Migration Inhibitory Factor (MIF) Promotes Cell Survival by Activation of the Akt Pathway and Role for CSN5/JAB1 in the Control of Autocrine MIF Activity. Oncogene 2007, 26, 5046–5059. [Google Scholar] [CrossRef]
- Lue, H.; Dewor, M.; Leng, L.; Bucala, R.; Bernhagen, J. Activation of the JNK Signalling Pathway by Macrophage Migration Inhibitory Factor (MIF) and Dependence on CXCR4 and CD74. Cell Signal 2011, 23, 135–144. [Google Scholar] [CrossRef]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage Migration Inhibitory Factor Induces B Cell Survival by Activation of a CD74-CD44 Receptor Complex. J. Biol. Chem. 2008, 283, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- De, R.; Sarkar, S.; Mazumder, S.; Debsharma, S.; Siddiqui, A.A.; Saha, S.J.; Banerjee, C.; Nag, S.; Saha, D.; Pramanik, S.; et al. Macrophage Migration Inhibitory Factor Regulates Mitochondrial Dynamics and Cell Growth of Human Cancer Cell Lines through CD74–NF-ΚB Signaling. J. Biol. Chem. 2018, 293, 19740–19760. [Google Scholar] [CrossRef] [PubMed]
- Streetly, M.J.; Maharaj, L.; Joel, S.; Schey, S.A.; Gribben, J.G.; Cotter, F.E. GCS-100, a Novel Galectin-3 Antagonist, Modulates MCL-1, NOXA, and Cell Cycle to Induce Myeloma Cell Death. Blood 2010, 115, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Cecchinelli, B.; Lavra, L.; Rinaldo, C.; Iacovelli, S.; Gurtner, A.; Gasbarri, A.; Ulivieri, A.; Del Prete, F.; Trovato, M.; Piaggio, G.; et al. Repression of the Antiapoptotic Molecule Galectin-3 by Homeodomain-Interacting Protein Kinase 2-Activated P53 Is Required for P53-Induced Apoptosis. Mol. Cell Biol. 2006, 26, 4746–4757. [Google Scholar] [CrossRef]
- Lavra, L.; Rinaldo, C.; Ulivieri, A.; Luciani, E.; Fidanza, P.; Giacomelli, L.; Bellotti, C.; Ricci, A.; Trovato, M.; Soddu, S.; et al. The Loss of the P53 Activator HIPK2 Is Responsible for Galectin-3 Overexpression in Well Differentiated Thyroid Carcinomas. PLoS ONE 2011, 6, e20665. [Google Scholar] [CrossRef]
- Raimond, J.; Rouleux, F.; Monsigny, M.; Legrand, A. The Second Intron of the Human Galectin-3 Gene Has a Strong Promoter Activity down-Regulated by P53. FEBS Lett. 1995, 363, 165–169. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, X.; Nash, G.B.; Stone, P.C.; Hilkens, J.; Rhodes, J.M.; Yu, L.-G. Circulating Galectin-3 Promotes Metastasis by Modifying MUC1 Localization on Cancer Cell Surface. Cancer Res. 2009, 69, 6799–6806. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor Suppressor P53: Biology, Signaling Pathways, and Therapeutic Targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-XL-Dependent Resistance to the BCL-2 Inhibitor ABT-199 Can Be Overcome by Preventing PI3K/AKT/MTOR Activation in Lymphoid Malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-Apoptotic Mcl-1 Is Essential for the Development and Sustained Growth of Acute Myeloid Leukemia. Genes. Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Akahani, S.; Nangia-Makker, P.; Inohara, H.; Kim, H.R.; Raz, A. Galectin-3: A Novel Antiapoptotic Molecule with a Functional BH1 (NWGR) Domain of Bcl-2 Family. Cancer Res. 1997, 57, 5272–5276. [Google Scholar] [PubMed]
- Harazono, Y.; Nakajima, K.; Raz, A. Why Anti-Bcl-2 Clinical Trials Fail: A Solution. Cancer Metastasis Rev. 2014, 33, 285–294. [Google Scholar] [CrossRef]
- Wu, Y.; Mehew, J.W.; Heckman, C.A.; Arcinas, M.; Boxer, L.M. Negative Regulation of Bcl-2 Expression by P53 in Hematopoietic Cells. Oncogene 2001, 20, 240–251. [Google Scholar] [CrossRef]
- Xu, J.; Dong, X.; Huang, D.C.S.; Xu, P.; Zhao, Q.; Chen, B. Current Advances and Future Strategies for BCL-2 Inhibitors: Potent Weapons against Cancers. Cancers 2023, 15, 4957. [Google Scholar] [CrossRef]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 Mediates Bone Marrow Microenvironment-Induced Drug Resistance in Acute Leukemia Cells via Wnt/β-Catenin Signaling Pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef]
- Song, S.; Mazurek, N.; Liu, C.; Sun, Y.; Ding, Q.Q.; Liu, K.; Hung, M.-C.; Bresalier, R.S. Galectin-3 Mediates Nuclear β-Catenin Accumulation and Wnt Signaling in Human Colon Cancer Cells by Regulation of Glycogen Synthase Kinase-3β Activity. Cancer Res. 2009, 69, 1343–1349. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shimura, T.; Yajima, T.; Kubo, N.; Araki, K.; Tsutsumi, S.; Suzuki, H.; Kuwano, H.; Raz, A. Transient Gene Silencing of Galectin-3 Suppresses Pancreatic Cancer Cell Migration and Invasion through Degradation of Β-catenin. Int. J. Cancer 2011, 129, 2775–2786. [Google Scholar] [CrossRef]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J.T. Wnt Signaling Strength Regulates Normal Hematopoiesis and Its Deregulation Is Involved in Leukemia Development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef]
- GE, X.; Wang, X. Role of Wnt Canonical Pathway in Hematological Malignancies. J. Hematol. Oncol. 2010, 3, 33. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/β-Catenin Pathway Is Required for the Development of Leukemia Stem Cells in AML. Science (1979) 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the β-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Lin, H.-M.; Pestell, R.G.; Raz, A.; Kim, H.-R.C. Galectin-3 Enhances Cyclin D(1) Promoter Activity through SP1 and a CAMP-Responsive Element in Human Breast Epithelial Cells. Oncogene 2002, 21, 8001–8010. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen Synthase Kinase-3 Regulates Mitochondrial Outer Membrane Permeabilization and Apoptosis by Destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute Myeloid Leukemia Transforms the Bone Marrow Niche into a Leukemia-Permissive Microenvironment through Exosome Secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Borgovan, T.; Crawford, L.; Nwizu, C.; Quesenberry, P. Stem Cells and Extracellular Vesicles: Biological Regulators of Physiology and Disease. Am. J. Physiol.-Cell Physiol. 2019, 317, C155–C166. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Kargar-sichani, Y.; Mohammadi, M.H.; Amiri, V.; Barzegar, M.; Keshavarz, A.; Bashash, D.; Farsani, M.A. Effect of Acute Myeloid Leukemia-Derived Extracellular Vesicles on Bone Marrow Mesenchymal Stromal Cells: Expression of Poor Prognosis Genes. Arch. Med. Res. 2023, 54, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, Y.; Shimada, H.; Wang, L.-C.; Zandi, E.; DeClerck, Y.A. Identification of Galectin-3-Binding Protein as a Factor Secreted by Tumor Cells That Stimulates Interleukin-6 Expression in the Bone Marrow Stroma. J. Biol. Chem. 2008, 283, 18573–18581. [Google Scholar] [CrossRef] [PubMed]
- Silverman, A.M.; Nakata, R.; Shimada, H.; Sposto, R.; DeClerck, Y.A. A Galectin-3–Dependent Pathway Upregulates Interleukin-6 in the Microenvironment of Human Neuroblastoma. Cancer Res. 2012, 72, 2228–2238. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogné, J.-M. Transfer of Multidrug Resistance among Acute Myeloid Leukemia Cells via Extracellular Vesicles and Their MicroRNA Cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Khandanpour, C.; Cadeddu, R.-P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional Inhibition of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Ruvolo, P.P.; Wang, R.-Y.; Battula, V.L.; Shpall, E.J.; Ruvolo, V.R.; McQueen, T.; Qui, Y.; Zeng, Z.; Pierce, S.; et al. Distinct Protein Signatures of Acute Myeloid Leukemia Bone Marrow-Derived Stromal Cells Are Prognostic for Patient Survival. Haematologica 2018, 103, 810–821. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-Catenin Signaling by Posttranslational Modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Ruvolo, V.R.; Burks, J.K.; Qiu, Y.; Wang, R.-Y.; Shpall, E.J.; Mirandola, L.; Hail, N.; Zeng, Z.; McQueen, T.; et al. Role of MSC-Derived Galectin 3 in the AML Microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 959–969. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis Induced by an Activating β-Catenin Mutation in Osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and Non-Canonical WNT Signaling in Cancer Stem Cells and Their Niches: Cellular Heterogeneity, Omics Reprogramming, Targeted Therapy and Tumor Plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef]
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/β-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2417–2429. [Google Scholar] [CrossRef]
- Shimura, T.; Takenaka, Y.; Tsutsumi, S.; Hogan, V.; Kikuchi, A.; Raz, A. Galectin-3, a Novel Binding Partner of Beta-Catenin. Cancer Res. 2004, 64, 6363–6367. [Google Scholar] [CrossRef]
- Shimura, T.; Takenaka, Y.; Fukumori, T.; Tsutsumi, S.; Okada, K.; Hogan, V.; Kikuchi, A.; Kuwano, H.; Raz, A. Implication of Galectin-3 in Wnt Signaling. Cancer Res. 2005, 65, 3535–3537. [Google Scholar] [CrossRef]
- Funasaka, T.; Raz, A.; Nangia-Makker, P. Nuclear Transport of Galectin-3 and Its Therapeutic Implications. Semin. Cancer Biol. 2014, 27, 30–38. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of Beta-Catenin by AKT Promotes Beta-Catenin Transcriptional Activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Schelker, R.C.; Iberl, S.; Müller, G.; Hart, C.; Herr, W.; Grassinger, J. TGF-Β1 and CXCL12 Modulate Proliferation and Chemotherapy Sensitivity of Acute Myeloid Leukemia Cells Co-Cultured with Multipotent Mesenchymal Stromal Cells. Hematology 2018, 23, 337–345. [Google Scholar] [CrossRef]
- Xu, Y.; Tabe, Y.; Jin, L.; Watt, J.; McQueen, T.; Ohsaka, A.; Andreeff, M.; Konopleva, M. TGF-β Receptor Kinase Inhibitor LY2109761 Reverses the Anti-apoptotic Effects of TGF-β1 in Myelo-monocytic Leukaemic Cells Co-cultured with Stromal Cells. Br. J. Haematol. 2008, 142, 192–201. [Google Scholar] [CrossRef]
- MacKinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of Transforming Growth Factor-Β1–Driven Lung Fibrosis by Galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Gong, D.; Shi, W.; Yi, S.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFβ Signaling Plays a Critical Role in Promoting Alternative Macrophage Activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Fei, C.; Guo, J.; Gu, S.; Zhu, Y.; Xu, F.; Zhang, Z.; Wu, L.; Li, X.; et al. Down-Regulation of Dicer1 Promotes Cellular Senescence and Decreases the Differentiation and Stem Cell-Supporting Capacities of Mesenchymal Stromal Cells in Patients with Myelodysplastic Syndrome. Haematologica 2015, 100, 194–204. [Google Scholar] [CrossRef]
- Li, Y.; Xu, X.; Wang, L.; Liu, G.; Li, Y.; Wu, X.; Jing, Y.; Li, H.; Wang, G. Senescent Mesenchymal Stem Cells Promote Colorectal Cancer Cells Growth via Galectin-3 Expression. Cell Biosci. 2015, 5, 21. [Google Scholar] [CrossRef]
- Mounayar, M.; Kefaloyianni, E.; Smith, B.; Solhjou, Z.; Maarouf, O.H.; Azzi, J.; Chabtini, L.; Fiorina, P.; Kraus, M.; Briddell, R.; et al. PI3kα and STAT1 Interplay Regulates Human Mesenchymal Stem Cell Immune Polarization. Stem Cells 2015, 33, 1892–1901. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-Induced Osteogenic Differentiation in Mesenchymal Stromal Cells Supports Leukemia Growth. JCI Insight 2017, 2, e90036. [Google Scholar] [CrossRef]
- Xu, L.; Qian, Z.; Wang, S.; Wang, R.; Pu, X.; Yang, B.; Zhou, Q.; Du, C.; Chen, Q.; Feng, Z.; et al. Galectin-3 Enhances Osteogenic Differentiation of Precursor Cells From Patients With Diffuse Idiopathic Skeletal Hyperostosis via Wnt/β-Catenin Signaling. J. Bone Miner. Res. 2022, 37, 724–739. [Google Scholar] [CrossRef]
- Weilner, S.; Keider, V.; Winter, M.; Harreither, E.; Salzer, B.; Weiss, F.; Schraml, E.; Messner, P.; Pietschmann, P.; Hildner, F.; et al. Vesicular Galectin-3 Levels Decrease with Donor Age and Contribute to the Reduced Osteo-Inductive Potential of Human Plasma Derived Extracellular Vesicles. Aging 2016, 8, 16–33. [Google Scholar] [CrossRef]
- Meng, Y.-B.; Li, X.; Li, Z.-Y.; Zhao, J.; Yuan, X.-B.; Ren, Y.; Cui, Z.-D.; Liu, Y.-D.; Yang, X.-J. MicroRNA-21 Promotes Osteogenic Differentiation of Mesenchymal Stem Cells by the PI3K/β-Catenin Pathway. J. Orthop. Res. 2015, 33, 957–964. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Gao, Q.; Xia, Y.; Liu, L.; Huang, L.; Liu, Y.; Zhang, X.; Xu, K.; Wei, J.; Hu, Y.; Mu, Y.; et al. Galectin-3 Enhances Migration of Minature Pig Bone Marrow Mesenchymal Stem Cells Through Inhibition of RhoA-GTP Activity. Sci. Rep. 2016, 6, 26577. [Google Scholar] [CrossRef]
- Sato, Y.; Mabuchi, Y.; Miyamoto, K.; Araki, D.; Niibe, K.; Houlihan, D.D.; Morikawa, S.; Nakagawa, T.; Nakajima, T.; Akazawa, C.; et al. Notch2 Signaling Regulates the Proliferation of Murine Bone Marrow-Derived Mesenchymal Stem/Stromal Cells via c-Myc Expression. PLoS ONE 2016, 11, e0165946. [Google Scholar] [CrossRef]
- Souza, B.S.d.F.; da Silva, K.N.; Silva, D.N.; Rocha, V.P.C.; Paredes, B.D.; Azevedo, C.M.; Nonaka, C.K.; Carvalho, G.B.; Vasconcelos, J.F.; dos Santos, R.R.; et al. Galectin-3 Knockdown Impairs Survival, Migration, and Immunomodulatory Actions of Mesenchymal Stromal Cells in a Mouse Model of Chagas Disease Cardiomyopathy. Stem Cells Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- An, L.; Chang, G.; Zhang, L.; Wang, P.; Gao, W.; Li, X. Pectin: Health-Promoting Properties as a Natural Galectin-3 Inhibitor. Glycoconj. J. 2024, 41, 93–118. [Google Scholar] [CrossRef]
- Zetterberg, F.R.; MacKinnon, A.; Brimert, T.; Gravelle, L.; Johnsson, R.E.; Kahl-Knutson, B.; Leffler, H.; Nilsson, U.J.; Pedersen, A.; Peterson, K.; et al. Discovery and Optimization of the First Highly Effective and Orally Available Galectin-3 Inhibitors for Treatment of Fibrotic Disease. J. Med. Chem. 2022, 65, 12626–12638. [Google Scholar] [CrossRef]
- Lepur, A.; Carlsson, M.C.; Novak, R.; Dumić, J.; Nilsson, U.J.; Leffler, H. Galectin-3 Endocytosis by Carbohydrate Independent and Dependent Pathways in Different Macrophage like Cell Types. Biochim. Biophys. Acta 2012, 1820, 804–818. [Google Scholar] [CrossRef]
- Hirani, N.; MacKinnon, A.C.; Nicol, L.; Ford, P.; Schambye, H.; Pedersen, A.; Nilsson, U.J.; Leffler, H.; Sethi, T.; Tantawi, S.; et al. Target Inhibition of Galectin-3 by Inhaled TD139 in Patients with Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2021, 57, 2002559. [Google Scholar] [CrossRef]
- Humphries, D.C.; Mills, R.; Boz, C.; McHugh, B.J.; Hirani, N.; Rossi, A.G.; Pedersen, A.; Schambye, H.T.; Slack, R.J.; Leffler, H.; et al. Galectin-3 Inhibitor GB0139 Protects against Acute Lung Injury by Inhibiting Neutrophil Recruitment and Activation. Front. Pharmacol. 2022, 13, 949264. [Google Scholar] [CrossRef]
- Chen, W.-S.; Cao, Z.; Leffler, H.; Nilsson, U.J.; Panjwani, N. Galectin-3 Inhibition by a Small-Molecule Inhibitor Reduces Both Pathological Corneal Neovascularization and Fibrosis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 9–20. [Google Scholar] [CrossRef]
- Comeglio, P.; Guarnieri, G.; Filippi, S.; Cellai, I.; Acciai, G.; Holyer, I.; Zetterberg, F.; Leffler, H.; Kahl-Knutson, B.; Sarchielli, E.; et al. The Galectin-3 Inhibitor Selvigaltin Reduces Liver Inflammation and Fibrosis in a High Fat Diet Rabbit Model of Metabolic-Associated Steatohepatitis. Front. Pharmacol. 2024, 15, 1430109. [Google Scholar] [CrossRef]
- Aslanis, V.; Abd-Elaziz, K.; Slack, R.J.; Brinch, A.; Gravelle, L.; Morley, W.; Phung, D.; Herman, K.; Holyer, I.; Poulsen, K.K.; et al. Relative Bioavailability and Food Effect of the Galectin-3 Inhibitor Selvigaltin (GB1211) Administered as a Tablet in Healthy Participants (GALBA-1). Cancer Chemother. Pharmacol. 2024, 94, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, P.; Zhang, Y.; Han, L.; Hu, Z.; Cai, Z.; Cai, J. Inhibition of Galectin-3 Augments the Antitumor Efficacy of PD-L1 Blockade in Non-small-cell Lung Cancer. FEBS Open Bio 2021, 11, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Mabbitt, J.; Holyer, I.D.; Roper, J.A.; Nilsson, U.J.; Zetterberg, F.R.; Vuong, L.; Mackinnon, A.C.; Pedersen, A.; Slack, R.J. Resistance to Anti-PD-1/Anti-PD-L1: Galectin-3 Inhibition with GB1211 Reverses Galectin-3-Induced Blockade of Pembrolizumab and Atezolizumab Binding to PD-1/PD-L1. Front. Immunol. 2023, 14, 1250559. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Lee, C.; Kim, H.-S.; Gu, S.J.; Yoon, H.J.; Won, S.B.; Lee, H.; Lee, Y.S.; Kim, S.S.; Kane, L.P.; et al. TIM-3 on Myeloid Cells Promotes Pulmonary Inflammation through Increased Production of Galectin-3. Commun. Biol. 2024, 7, 1090. [Google Scholar] [CrossRef]
- Yu, L.-G.; Andrews, N.; Zhao, Q.; McKean, D.; Williams, J.F.; Connor, L.J.; Gerasimenko, O.V.; Hilkens, J.; Hirabayashi, J.; Kasai, K.; et al. Galectin-3 Interaction with Thomsen-Friedenreich Disaccharide on Cancer-Associated MUC1 Causes Increased Cancer Cell Endothelial Adhesion. J. Biol. Chem. 2007, 282, 773–781. [Google Scholar] [CrossRef]
- Tanida, S.; Mori, Y.; Ishida, A.; Akita, K.; Nakada, H. Galectin-3 Binds to MUC1-N-Terminal Domain and Triggers Recruitment of β-Catenin in MUC1-Expressing Mouse 3T3 Cells. Biochim. Biophys. Acta 2014, 1840, 1790–1797. [Google Scholar] [CrossRef]
- Piyush, T.; Chacko, A.R.; Sindrewicz, P.; Hilkens, J.; Rhodes, J.M.; Yu, L.-G. Interaction of Galectin-3 with MUC1 on Cell Surface Promotes EGFR Dimerization and Activation in Human Epithelial Cancer Cells. Cell Death Differ. 2017, 24, 1937–1947. [Google Scholar] [CrossRef]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of Cytokine Receptors by Golgi N-Glycan Processing and Endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef]
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 Protein Modulates Cell Surface Expression and Activation of Vascular Endothelial Growth Factor Receptor 2 in Human Endothelial Cells. J. Biol. Chem. 2011, 286, 29913–29921. [Google Scholar] [CrossRef]
- Jastrzębski, K.; Zdżalik-Bielecka, D.; Mamińska, A.; Kalaidzidis, Y.; Hellberg, C.; Miaczynska, M. Multiple Routes of Endocytic Internalization of PDGFRβ Contribute to PDGF-Induced STAT3 Signaling. J. Cell Sci. 2017, 130, 577–589. [Google Scholar] [CrossRef]
- Rosenberg, I.; Cherayil, B.J.; Isselbacher, K.J.; Pillai, S. Mac-2-Binding Glycoproteins. Putative Ligands for a Cytosolic Beta-Galactoside Lectin. J. Biol. Chem. 1991, 266, 18731–18736. [Google Scholar] [CrossRef] [PubMed]
- Inohara, H.; Akahani, S.; Koths, K.; Raz, A. Interactions between Galectin-3 and Mac-2-Binding Protein Mediate Cell-Cell Adhesion. Cancer Res. 1996, 56, 4530–4534. [Google Scholar] [PubMed]
- Sasaki, T.; Brakebusch, C.; Engel, J.; Timpl, R. Mac-2 Binding Protein Is a Cell-Adhesive Protein of the Extracellular Matrix Which Self-Assembles into Ring-like Structures and Binds Beta1 Integrins, Collagens and Fibronectin. EMBO J. 1998, 17, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Boscher, C.; Zheng, Y.Z.; Lakshminarayan, R.; Johannes, L.; Dennis, J.W.; Foster, L.J.; Nabi, I.R. Galectin-3 Protein Regulates Mobility of N-Cadherin and GM1 Ganglioside at Cell-Cell Junctions of Mammary Carcinoma Cells. J. Biol. Chem. 2012, 287, 32940–32952. [Google Scholar] [CrossRef]
- Markowska, A.I.; Liu, F.-T.; Panjwani, N. Galectin-3 Is an Important Mediator of VEGF- and BFGF-Mediated Angiogenic Response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An Integrin β3-KRAS-RalB Complex Drives Tumour Stemness and Resistance to EGFR Inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef]
- Clark, M.C.; Pang, M.; Hsu, D.K.; Liu, F.-T.; de Vos, S.; Gascoyne, R.D.; Said, J.; Baum, L.G. Galectin-3 Binds to CD45 on Diffuse Large B-Cell Lymphoma Cells to Regulate Susceptibility to Cell Death. Blood 2012, 120, 4635–4644. [Google Scholar] [CrossRef]
- Lakshminarayan, R.; Wunder, C.; Becken, U.; Howes, M.T.; Benzing, C.; Arumugam, S.; Sales, S.; Ariotti, N.; Chambon, V.; Lamaze, C.; et al. Galectin-3 Drives Glycosphingolipid-Dependent Biogenesis of Clathrin-Independent Carriers. Nat. Cell Biol. 2014, 16, 595–606. [Google Scholar] [CrossRef]
- Priglinger, C.S.; Szober, C.M.; Priglinger, S.G.; Merl, J.; Euler, K.N.; Kernt, M.; Gondi, G.; Behler, J.; Geerlof, A.; Kampik, A.; et al. Galectin-3 Induces Clustering of CD147 and Integrin-Β1 Transmembrane Glycoprotein Receptors on the RPE Cell Surface. PLoS ONE 2013, 8, e70011. [Google Scholar] [CrossRef]
- Feuk-Lagerstedt, E.; Jordan, E.T.; Leffler, H.; Dahlgren, C.; Karlsson, A. Identification of CD66a and CD66b as the Major Galectin-3 Receptor Candidates in Human Neutrophils. J. Immunol. 1999, 163, 5592–5598. [Google Scholar] [CrossRef]
- MacKinnon, A.C.; Farnworth, S.L.; Hodkinson, P.S.; Henderson, N.C.; Atkinson, K.M.; Leffler, H.; Nilsson, U.J.; Haslett, C.; Forbes, S.J.; Sethi, T. Regulation of Alternative Macrophage Activation by Galectin-3. J. Immunol. 2008, 180, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, C.; Meng, J.; Li, N.; Xu, Z.; Liu, X.; Hou, S. Galectin-3 Regulates Microglial Activation and Promotes Inflammation through TLR4/MyD88/NF-KB in Experimental Autoimmune Uveitis. Clin. Immunol. 2022, 236, 108939. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hsu, D.K.; Chen, H.-Y.; Yang, R.-Y.; Carraway, K.L.; Isseroff, R.R.; Liu, F.-T. Galectin-3 Regulates Intracellular Trafficking of EGFR through Alix and Promotes Keratinocyte Migration. J. Investig. Dermatol. 2012, 132, 2828–2837. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakai, T.; Sano, N.; Fukui, K. Nucling Mediates Apoptosis by Inhibiting Expression of Galectin-3 through Interference with Nuclear Factor KappaB Signalling. Biochem. J. 2004, 380, 31–41. [Google Scholar] [CrossRef]
- Jia, W.; Kong, L.; Kidoya, H.; Naito, H.; Muramatsu, F.; Hayashi, Y.; Hsieh, H.-Y.; Yamakawa, D.; Hsu, D.K.; Liu, F.-T.; et al. Indispensable Role of Galectin-3 in Promoting Quiescence of Hematopoietic Stem Cells. Nat. Commun. 2021, 12, 2118. [Google Scholar] [CrossRef]
- Ma, J.; Yao, Y.; Wang, P.; Liu, Y.; Zhao, L.; Li, Z.; Li, Z.; Xue, Y. MiR-152 Functions as a Tumor Suppressor in Glioblastoma Stem Cells by Targeting Krüppel-like Factor 4. Cancer Lett. 2014, 355, 85–95. [Google Scholar] [CrossRef]
- Yıldırım, C. Galectin-9, a pro-Survival Factor Inducing Immunosuppression, Leukemic Cell Transformation and Expansion. Mol. Biol. Rep. 2024, 51, 571. [Google Scholar] [CrossRef]
- Balan, V.; Nangia-Makker, P.; Schwartz, A.G.; Jung, Y.S.; Tait, L.; Hogan, V.; Raz, T.; Wang, Y.; Yang, Z.Q.; Wu, G.S.; et al. Racial Disparity in Breast Cancer and Functional Germ Line Mutation in Galectin-3 (Rs4644): A Pilot Study. Cancer Res. 2008, 68, 10045–10050. [Google Scholar] [CrossRef]
- Matarrese, P.; Fusco, O.; Tinari, N.; Natoli, C.; Liu, F.T.; Semeraro, M.L.; Malorni, W.; Iacobelli, S. Galectin-3 Overexpression Protects from Apoptosis by Improving Cell Adhesion Properties. Int. J. Cancer 2000, 85, 545–554. [Google Scholar] [CrossRef]
- Sioud, M. New Insights into Mesenchymal Stromal Cell-Mediated T-Cell Suppression through Galectins. Scand. J. Immunol. 2011, 73, 79–84. [Google Scholar] [CrossRef]
- Stillman, B.N.; Hsu, D.K.; Pang, M.; Brewer, C.F.; Johnson, P.; Liu, F.-T.; Baum, L.G. Galectin-3 and Galectin-1 Bind Distinct Cell Surface Glycoprotein Receptors to Induce T Cell Death. J. Immunol. 2006, 176, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Qian, Y.; Karmakar, S.; Koyama, N.S.; Dias-Baruffi, M.; Leffler, H.; McEver, R.P.; Cummings, R.D. Differential Roles of Galectin-1 and Galectin-3 in Regulating Leukocyte Viability and Cytokine Secretion. J. Immunol. 2008, 180, 3091–3102. [Google Scholar] [CrossRef]
- Demotte, N.; Wieërs, G.; Van Der Smissen, P.; Moser, M.; Schmidt, C.; Thielemans, K.; Squifflet, J.-L.; Weynand, B.; Carrasco, J.; Lurquin, C.; et al. A Galectin-3 Ligand Corrects the Impaired Function of Human CD4 and CD8 Tumor-Infiltrating Lymphocytes and Favors Tumor Rejection in Mice. Cancer Res. 2010, 70, 7476–7488. [Google Scholar] [CrossRef] [PubMed]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef]
- Sioud, M.; Mobergslien, A.; Boudabous, A.; Fløisand, Y. Evidence for the Involvement of Galectin-3 in Mesenchymal Stem Cell Suppression of Allogeneic T-Cell Proliferation. Scand. J. Immunol. 2010, 71, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-Released Galectin-3, a Soluble Inhibitory Ligand of Human NKp30, Plays an Important Role in Tumor Escape from NK Cell Attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef]
- Tsuboi, S.; Sutoh, M.; Hatakeyama, S.; Hiraoka, N.; Habuchi, T.; Horikawa, Y.; Hashimoto, Y.; Yoneyama, T.; Mori, K.; Koie, T.; et al. A Novel Strategy for Evasion of NK Cell Immunity by Tumours Expressing Core2 O-Glycans. EMBO J. 2011, 30, 3173–3185. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sutoh, M.; Hatakeyama, S.; Mori, K.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Habuchi, T.; et al. MUC1 Carrying Core 2 O-Glycans Functions as a Molecular Shield against NK Cell Attack, Promoting Bladder Tumor Metastasis. Int. J. Oncol. 2012, 40, 1831–1838. [Google Scholar] [CrossRef]
- Jia, W.; Kidoya, H.; Yamakawa, D.; Naito, H.; Takakura, N. Galectin-3 Accelerates M2 Macrophage Infiltration and Angiogenesis in Tumors. Am. J. Pathol. 2013, 182, 1821–1831. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Raz, T.; Tait, L.; Hogan, V.; Fridman, R.; Raz, A. Galectin-3 Cleavage: A Novel Surrogate Marker for Matrix Metalloproteinase Activity in Growing Breast Cancers. Cancer Res. 2007, 67, 11760–11768. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, Z.; Liu, S.; Dong, Z.; Dalin, M.; Bao, S.; Hu, Y.; Wei, F. Galectin-3 Gene Silencing Inhibits Migration and Invasion of Human Tongue Cancer Cells in Vitro via Downregulating β-Catenin. Acta Pharmacol. Sin. 2013, 34, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Brandwein, J.; Yi, Q.-L.; Chun, K.; Patterson, B.; Brien, B. Extramedullary Infiltrates of AML Are Associated with CD56 Expression, 11q23 Abnormalities and Inferior Clinical Outcome. Leuk. Res. 2004, 28, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, R.; Tawa, A.; Hanada, R.; Horibe, K.; Tsuchida, M.; Tsukimoto, I.; Japanese Childhood AML Cooperative Study Group. Extramedullary Infiltration at Diagnosis and Prognosis in Children with Acute Myelogenous Leukemia. Pediatr. Blood Cancer 2007, 48, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, J.P.; Orudjev, E.; Bunin, N.; Felix, C.A.; Lange, B.J. Isolated Extramedullary Relapse in Acute Myeloid Leukemia: A Retrospective Analysis. Med. Pediatr. Oncol. 2002, 38, 387–390. [Google Scholar] [CrossRef]
- Eckardt, J.-N.; Stölzel, F.; Kunadt, D.; Röllig, C.; Stasik, S.; Wagenführ, L.; Jöhrens, K.; Kuithan, F.; Krämer, A.; Scholl, S.; et al. Molecular Profiling and Clinical Implications of Patients with Acute Myeloid Leukemia and Extramedullary Manifestations. J. Hematol. Oncol. 2022, 15, 60. [Google Scholar] [CrossRef]
- Haase, R.; Wiegand, P.; Hirsch, W.; Meyer-Bahlburg, A.; Diwan, O.; Wawer, A.; Burdach, S. Unusual Presentation of Central Nervous System Relapse with Oculomotor Nerve Palsy in a Case of CD56-Positive Acute Myeloid Leukemia Following Allogeneic Stem Cell Transplantation. Pediatr. Transplant. 2002, 6, 260–265. [Google Scholar] [CrossRef]
- Chandra, D.J.; Alber, B.; Saultz, J.N. The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers 2024, 16, 2615. [Google Scholar] [CrossRef]
- Moore, C.G.; Stein, A.; Fathi, A.T.; Pullarkat, V. Treatment of Relapsed/Refractory AML-Novel Treatment Options Including Immunotherapy. Am. J. Hematol. 2025, 100 (Suppl. 2), 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bawek, S.; Gurusinghe, S.; Burwinkel, M.; Przespolewski, A. Updates in Novel Immunotherapeutic Strategies for Relapsed/Refractory AML. Front. Oncol. 2024, 14, 1374963. [Google Scholar] [CrossRef]
- Forsberg, M.; Konopleva, M. AML Treatment: Conventional Chemotherapy and Emerging Novel Agents. Trends Pharmacol. Sci. 2024, 45, 430–448. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Model/Cell Type | Drug | Effects | Study Type | Reference |
|---|---|---|---|---|
| THP-1 (M0, M1, M2), HFL-1, SKBR3 cell lines | TD139 | Binding and endocytosis of Gal-3 is inhibited in a carbohydrate dependent manner in non-macrophage cells and M2 macrophages. | Preclinical | [156] |
| Healthy subjects and IPF patients | TD139 | TD139 was safe and well tolerated in healthy subjects and IPF patients. TD139 reduced Gal-3 expression in alveolar macrophages and plasma biomarkers associated with IPF progression (PDGF-BB, PAI-1, Gal-3, CCL-18, and YKL-40). | Phase 1/2a clinical trial (NCT02257177) | [157] |
| Primary alveolar epithelial cells from WT mice (in vitro), mouse model of TGF-β1-induced lung fibrosis (in vivo) | TD139 | TD139 inhibited TGF-β1-induced β-catenin activation in vitro and in vivo. | Preclinical | [141] |
| Mouse model of bleomycin-induced lung fibrosis | TD139 | TD139 diminished lung fibrosis in vivo. | Preclinical | [141] |
| Peripheral human neutrophils and monocytes, and cell lines: THP-1, Jurkat E6, A549 | GB0139 | GB0139 inhibits inflammation in vitro by reducing neutrophil activation, monocyte IL-8 secretion, pro-inflammatory M1 macrophage activation, T cell apoptosis, and pro-inflammatory genes (e.g., IL-6, IL-8, TNF-α) in injured alveolar epithelial cells. | Preclinical | [158] |
| Mouse model of LPS/bleomycin-induced ALI | GB0139 | GB0139 reduces the progression of ALI in vivo by reducing the recruitment and activation of inflammatory cells, the activation of pro-inflammatory M1 macrophages, pro-inflammatory cytokines (e.g., IL-6 and TNF-α) and pro-fibrotic cytokines (e.g., MIP-1α) in BALf, while increasing cytotoxic CD3+CD8+ T cells. | Preclinical | [158] |
| Human umbilical vein endothelial cells (HUVECs) | TD139 | TD139 reduces VEGF-A-induced endothelial cell migration and sprouting in vitro. | Preclinical | [159] |
| Mouse models of silver nitrate cautery and alkaline burn injury | TD139 | TD139 inhibits corneal angiogenesis and fibrosis in vivo. | Preclinical | [159] |
| THP-1, LX2 cell lines | GB1211 | GB1211 inhibits cell surface Gal-3 expression in human macrophages and TGF-β-induced pro-fibrotic gene expression in human hepatic stellate cells in vitro. | Preclinical | [155] |
| Mouse models of CCl4-induced liver fibrosis and bleomycin-induced lung fibrosis | GB1211 | GB1211 exhibits anti-fibrotic activity in the liver and lungs in vivo. | Preclinical | [155] |
| HFD rabbit model of MASH | GB1211 | GB1211 decreased Gal-3 mRNA and protein levels in the liver. GB1211 reduced liver inflammation (e.g., infiltrates of macrophages), fibrosis (e.g., collagen deposition), biomarkers of liver disease (AST, ALT, bilirubin), and markers of inflammation and fibrosis at mRNA and protein levels (e.g., IL6, collagen, TGF-β3, SNAI2). | Preclinical | [160] |
| Healthy subjects | GB1211 | The tablet form of GB1211 had higher bioavailability than the capsule form. Food intake had minimal effects on the pharmacokinetics of the tablet form. 100 mg tablet form was well tolerated and did not cause serious side effects. | Phase 1 clinical trial (GALBA-1; NCT05747573). | [161] |
| A549 cell line, PBMCs | GB1107, Anti-PD-L1 | GB1107 decreased both PD-L1 expression and STAT3 phosphorylation in lung cancer cells in vitro. GB1107 enhanced the cytotoxic activity of T cells against lung cancer cells induced by PD-L1 blockade in vitro. | Preclinical | [162] |
| Mouse xenograft model of lung cancer | GB1107, Anti-PD-L1 | Combination of PD-L1 blockade and GB1107 synergistically reduced tumor growth in vivo, which was accompanied by increased infiltration of CD3+ TILs and granzyme B release into tumors. | Preclinical | [162] |
| Jurkat-Lucia™ TCR-hPD-1, Raji-APC-hPD-L1 cell lines | GB1211, Atezolizumab (anti-PD-L1), Pembrolizumab (anti-PD-1) | GB1211 reduced the binding of Gal-3 to both PD-1 and PD-L1, and restored the binding of atezolizumab and pembrolizumab to PD-L1 and PD-1, respectively. | Preclinical | [163] |
| LLC1 syngeneic mouse lung cancer model | GB1211, Atezolizumab (anti-PD-L1) | Combination treatment with GB1211 and anti-PD-L1 monoclonal antibody reduced tumor growth in vivo, which was accompanied by an increased percentage of CD8+ TILs. | Preclinical | [163] |
| FSF-TIM3/LysM-Cre+/− mouse model (Myeloid cells with TIM-3 overexpression) | GB1107, Anti-TIM-3 | TIM-3 blockade or GB1107 reduced lung inflammation in vivo. | Preclinical | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yıldırım, C. Galectin-3 Release in the Bone Marrow Microenvironment Promotes Drug Resistance and Relapse in Acute Myeloid Leukemia. Life 2025, 15, 937. https://doi.org/10.3390/life15060937
Yıldırım C. Galectin-3 Release in the Bone Marrow Microenvironment Promotes Drug Resistance and Relapse in Acute Myeloid Leukemia. Life. 2025; 15(6):937. https://doi.org/10.3390/life15060937
Chicago/Turabian StyleYıldırım, Cansu. 2025. "Galectin-3 Release in the Bone Marrow Microenvironment Promotes Drug Resistance and Relapse in Acute Myeloid Leukemia" Life 15, no. 6: 937. https://doi.org/10.3390/life15060937
APA StyleYıldırım, C. (2025). Galectin-3 Release in the Bone Marrow Microenvironment Promotes Drug Resistance and Relapse in Acute Myeloid Leukemia. Life, 15(6), 937. https://doi.org/10.3390/life15060937