
Genomic Insights and Comparative Analysis of Novel Rhodopseudomonas Species: A Purple Non-Sulfur Bacterium Isolated from Latex Rubber Sheet Wastewater

 , , , , , ,
, , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Bacterial Strains and Isolation of Genomic DNA
2.2. Whole-Genome Sequencing
2.3. Sequence Analysis and Visualization
2.4. 16S rRNA Phylogeny and Multilocus Sequence Analysis
2.5. Comparative Genome Analysis
2.6. Wastewater Treatment-Related Genes
3. Results and Discussion
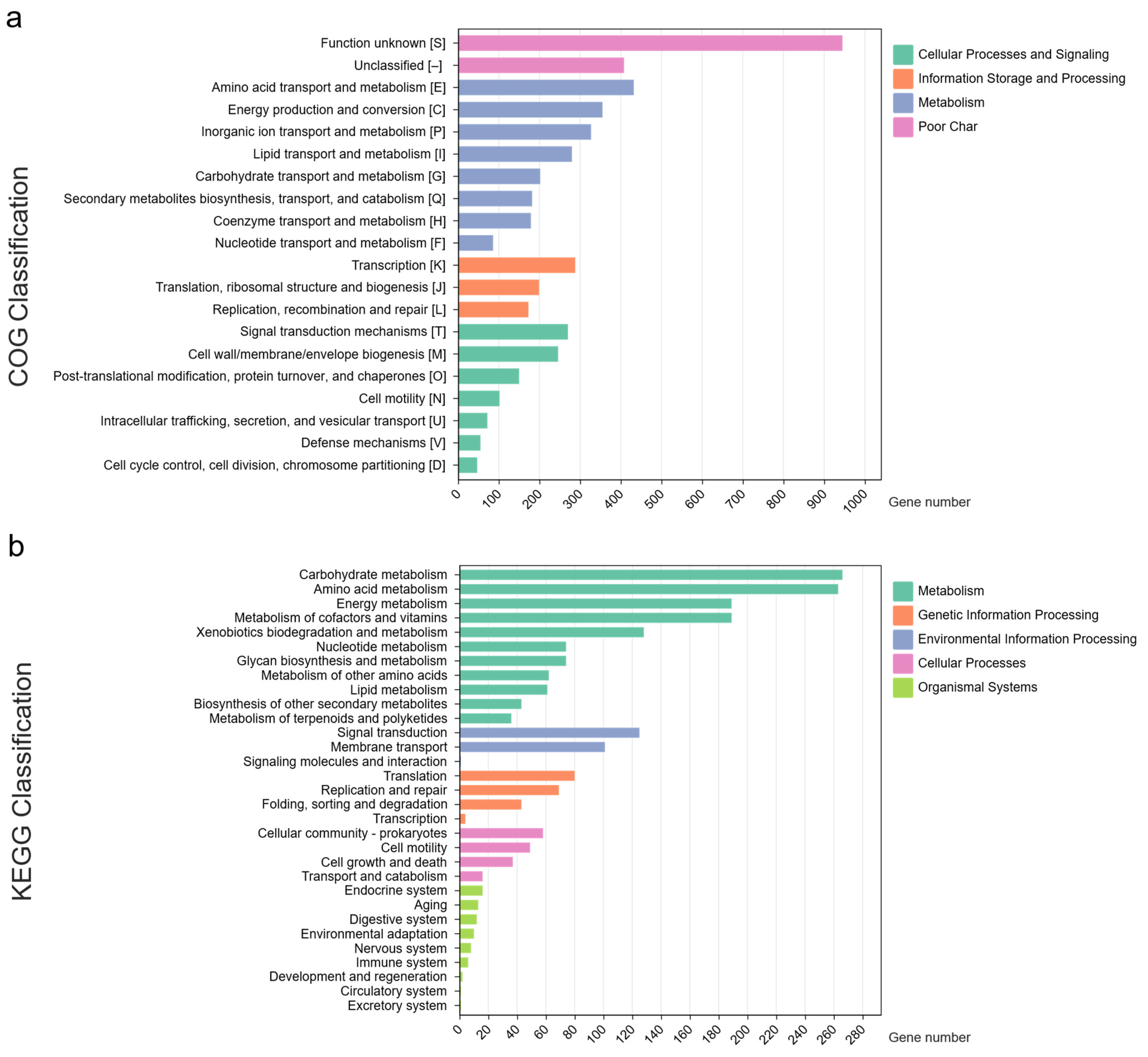
3.1. Genome Profiles of Novel Species
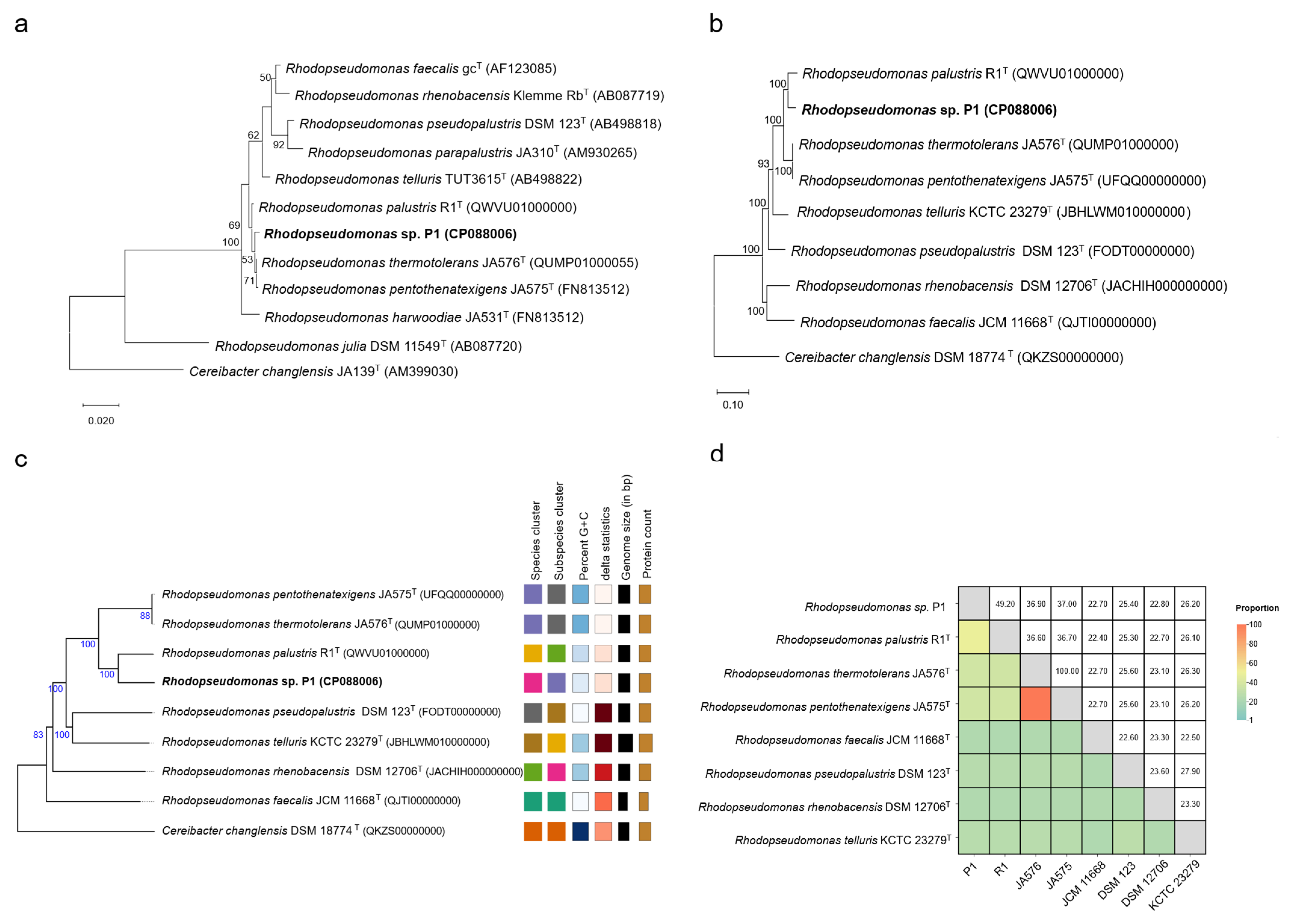
3.2. Phylogenetic Relationship Analysis
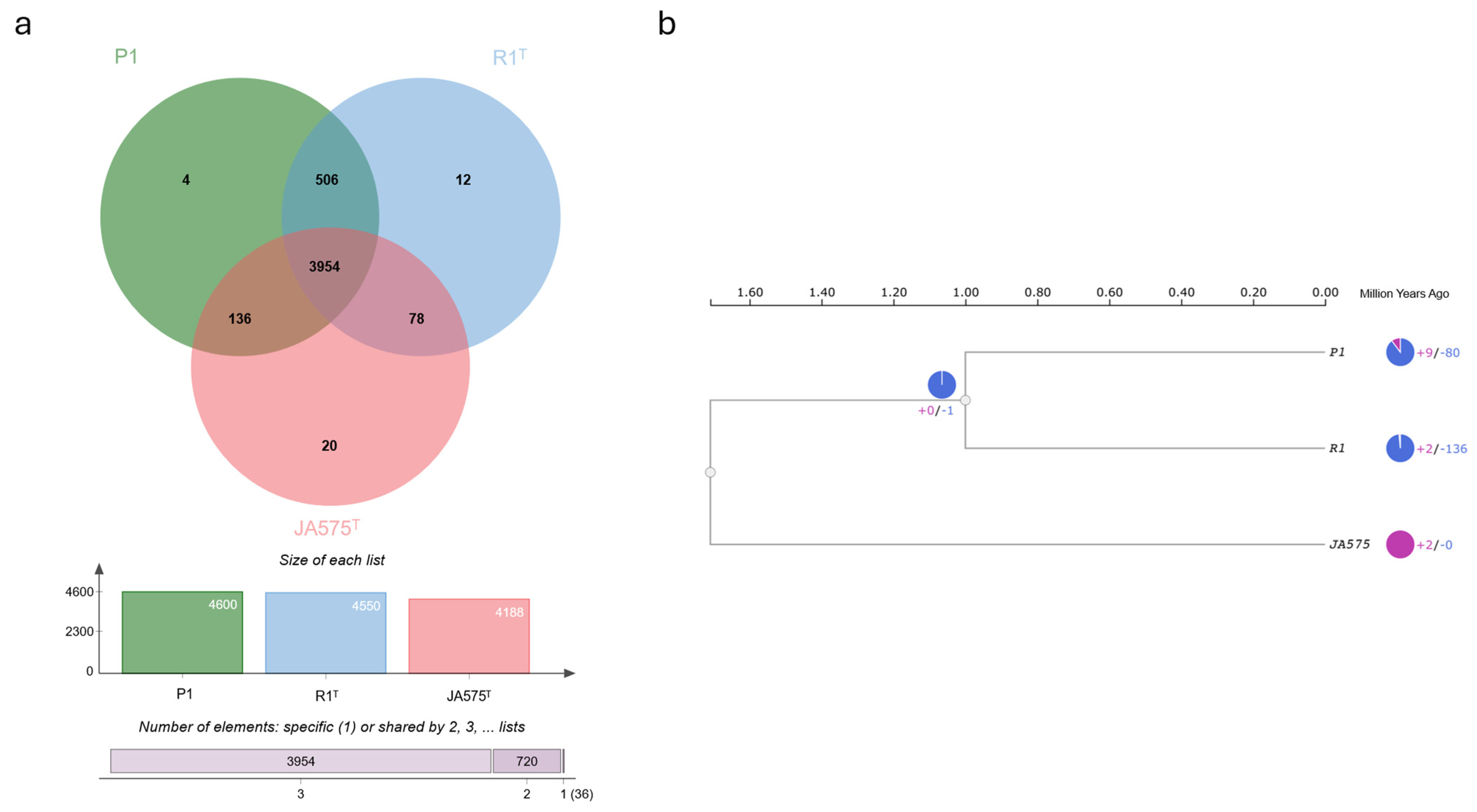
3.3. Comparative Genome Analysis of Novel Species
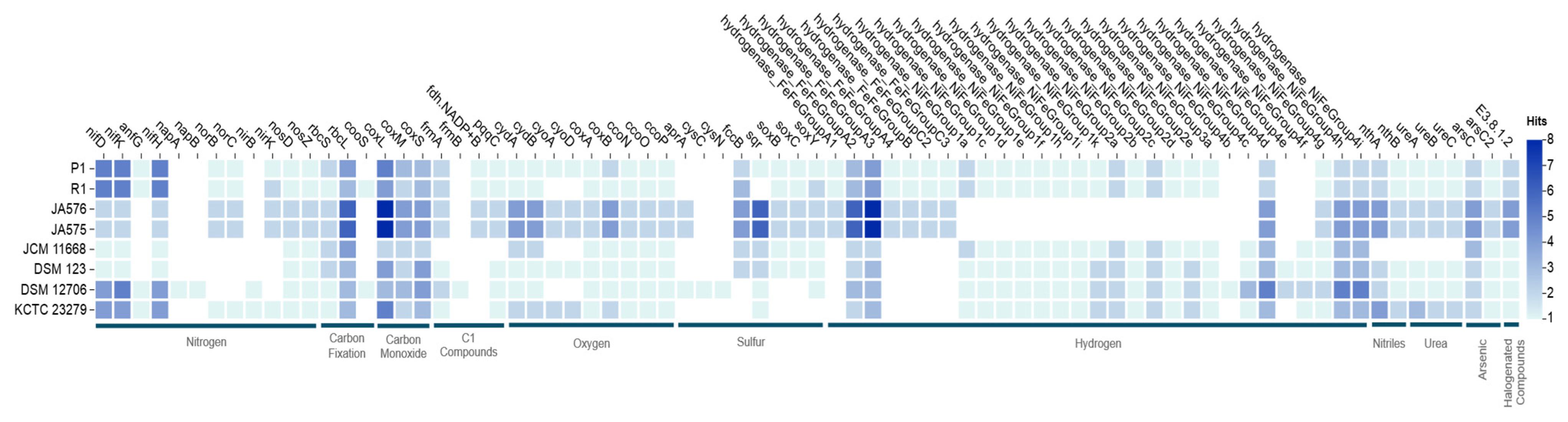
3.4. Bioelement Cycling and Metal Resistance in Rhodopseudomonas sp. P1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, L.-Y.; Jin, D.-C.; Zuo, H.; Zhang, Z.; Tan, X.-Q.; Zhang, D.-Y.; Lu, X.-Y.; Liu, Y. Effects of Rhodopseudomonas palustris PSB06 on pepper rhizosphere microbial community structure. Huan Jing Ke Xue 2017, 38, 735–742. [Google Scholar] [PubMed]
- Oda, Y.; Nelson, W.C.; Alexander, W.G.; Nguyen, S.; Egbert, R.G.; Harwood, C.S. A Rhodopseudomonas strain with a substantially smaller genome retains the core metabolic versatility of its genus. Appl. Environ. Microbiol. 2025, 91, e02056-24. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Doerner, K.F.; Alok, P.C.; Choudhary, M. Skatole remediation potential of Rhodopseudomonas palustris WKU-KDNS3 isolated from an animal waste lagoon. Lett. Appl. Microbiol. 2015, 60, 298–306. [Google Scholar] [CrossRef]
- Haq, I.U.; Christensen, A.; Fixen, K.R. Evolution of Rhodopseudomonas palustris to degrade halogenated aromatic compounds involves changes in pathway regulation and enzyme specificity. Appl. Environ. Microbiol. 2024, 90, e02104-23. [Google Scholar] [CrossRef]
- Yamanaka, K.; Tsuyuki, Y. Occurrence of dehydrogenases for the metabolism of vanillyl alcohol in Rhodopseudomonas acidophila M402. Agric. Biol. Chem. 1983, 47, 1361–1362. [Google Scholar] [CrossRef]
- Mutharasaiah, K.; Govindareddy, V.; Chandrakant, K. Biodegradation of 2-chlorophenol by Rhodopseudomonas palustris. Bioremediat. J. 2012, 16, 1–8. [Google Scholar] [CrossRef]
- Li, M.; Ning, P.; Sun, Y.; Luo, J.; Yang, J. Characteristics and application of Rhodopseudomonas palustris as a microbial cell factory. Front. Bioeng. Biotechnol. 2022, 10, 897003. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, Y.; Li, X. Removal of pollutants and accumulation of high-value cell inclusions in a batch reactor containing Rhodopseudomonas for treating real heavy oil refinery wastewater. J. Environ. Manag. 2023, 345, 118834. [Google Scholar] [CrossRef]
- Harwood, C.S.; Gibson, J. Anaerobic and aerobic metabolism of diverse aromatic compounds by the photosynthetic bacterium Rhodopseudomonas palustris. Appl. Environ. Microbiol. 1988, 54, 712–717. [Google Scholar] [CrossRef]
- Kornochalert, N.; Kantachote, D.; Chaiprapat, S.; Techkarnjanaruk, S. Use of Rhodopseudomonas palustris P1 stimulated growth by fermented pineapple extract to treat latex rubber sheet wastewater to obtain single cell protein. Ann. Microbiol. 2014, 64, 1021–1032. [Google Scholar] [CrossRef]
- Oda, Y.; Larimer, F.W.; Chain, P.S.; Malfatti, S.; Shin, M.V.; Vergez, L.M.; Hauser, L.; Land, M.L.; Braatsch, S.; Beatty, J.T. Multiple genome sequences reveal adaptations of a phototrophic bacterium to sediment microenvironments. Proc. Natl. Acad. Sci. USA 2008, 105, 18543–18548. [Google Scholar] [CrossRef] [PubMed]
- Kantachote, D.; Torpee, S.; Umsakul, K. The potential use of anoxygenic phototrophic bacteria for treating latex rubber sheet wastewater. Electron. J. Biotechnol. 2005, 8. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W. Introducing the bacterial and viral bioinformatics resource center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Group, S.F.U.R.C.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder-distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Chivian, D.; Jungbluth, S.P.; Dehal, P.S.; Wood-Charlson, E.M.; Canon, R.S.; Allen, B.H.; Clark, M.M.; Gu, T.; Land, M.L.; Price, G.A. Metagenome-assembled genome extraction and analysis from microbiomes using KBase. Nat. Protoc. 2023, 18, 208–238. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Alanjary, M.; Steinke, K.; Ziemert, N. AutoMLST: An automated web server for generating multi-locus species trees highlighting natural product potential. Nucleic Acids Res. 2019, 47, W276–W282. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- Choo, S.W.; Rishik, S.; Wee, W.Y. Comparative genome analyses of Mycobacteroides immunogenum reveals two potential novel subspecies. Microb. Genom. 2020, 6, e000495. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing cultivation to identify bacterial species. Microbe 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Cabal, A.; Jun, S.-R.; Jenjaroenpun, P.; Wanchai, V.; Nookaew, I.; Wongsurawat, T.; Burgess, M.J.; Kothari, A.; Wassenaar, T.M.; Ussery, D.W. Genome-based comparison of Clostridioides difficile: Average amino acid identity analysis of core genomes. Microb. Ecol. 2018, 76, 801–813. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- Nevers, Y.; Jones, T.E.; Jyothi, D.; Yates, B.; Ferret, M.; Portell-Silva, L.; Codo, L.; Cosentino, S.; Marcet-Houben, M.; Vlasova, A. The Quest for orthologs orthology benchmark service in 2022. Nucleic Acids Res. 2022, 50, W623–W632. [Google Scholar] [CrossRef] [PubMed]
- Karaoz, U.; Brodie, E.L. microTrait: A toolset for a trait-based representation of microbial genomes. Front. Bioinform. 2022, 2, 918853. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein families for the microbial genomes in the PATRIC database. Front. Microbiol. 2016, 7, 118. [Google Scholar] [CrossRef]
- Lopez-Romero, J.; Salgado-Manjarrez, E.; Torres, L.; Garcia-Peña, E.I. Enhanced carotenoid production by Rhodopseudomonas palustris ATCC 17001 under low light conditions. J. Biotechnol. 2020, 323, 159–165. [Google Scholar] [CrossRef]
- Li, M.; Zhu, T.; Yang, R.; Wang, Z.; Liu, M.; Yang, J. Carotenoids synthesis affects the salt tolerance mechanism of Rhodopseudomonas palustris. Front. Microbiol. 2023, 14, 1292937. [Google Scholar] [CrossRef]
- Saejung, C.; Ampornpat, W. Production and nutritional performance of carotenoid-producing photosynthetic bacterium Rhodopseudomonas faecalis PA2 grown in domestic wastewater intended for animal feed production. Waste Biomass Valorization 2019, 10, 299–310. [Google Scholar] [CrossRef]
- Huang, N.; Wang, Z.; Xiao, X.; Gai, T.; Zhao, D.; Liu, L.; Wu, W. Utilizing microbial electrochemical methods to enhance lycopene production in Rhodopseudomonas palustris. Foods 2024, 13, 3811. [Google Scholar] [CrossRef]
- Fujimoto, A.; Wong, J.H.; Yoshii, Y.; Akiyama, S.; Tanaka, A.; Yagi, H.; Shigemizu, D.; Nakagawa, H.; Mizokami, M.; Shimada, M. Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer. Genome Med. 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M.; Spröer, C.; Klenk, H.-P. When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Cantera, J.J.L.; Kawasaki, H.; Seki, T. The nitrogen-fixing gene (nifH) of Rhodopseudomonas palustris: A case of lateral gene transfer? Microbiology 2004, 150, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Huang, Y.Y.; Khan, Q.; Zhang, K.K.; Guo, D.J.; Yang, L.T.; Li, Y.R.; Xing, Y.X. Cloning, Prokaryotic expression and functional characterization of nifh gene from the associative nitrogen-fixing bacteria Klebsiella variicola DX120E. Iran J. Biotechnol. 2023, 21, e3451. [Google Scholar]
- Luo, D.; Chen, Q.; Jiang, B.; Lin, S.; Peng, L.; Zeng, L.; Hu, X.; Chen, K. Combination of dynamic turbidimetry and tube agglutination to identify procoagulant genes by transposon mutagenesis in Staphylococcus aureus. bioRxiv 2018, 426783. [Google Scholar] [CrossRef]
- Wodara, C.; Kostka, S.; Egert, M.; Kelly, D.P.; Friedrich, C.G. Identification and sequence analysis of the soxB gene essential for sulfur oxidation of Paracoccus denitrificans GB17. J. Bacteriol. 1994, 176, 6188–6191. [Google Scholar] [CrossRef]
- Jia, J.; Wang, Z.; Zhang, M.; Huang, C.; Song, Y.; Xu, F.; Zhang, J.; Li, J.; He, M.; Li, Y.; et al. SQR mediates therapeutic effects of H(2)S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci. Adv. 2020, 6, eaaz5752. [Google Scholar] [CrossRef]
- Miki, K.; Atomi, H.; Watanabe, S. Structural insight into [NiFe] hydrogenase maturation by transient complexes between hyp proteins. Acc. Chem. Res. 2020, 53, 875–886. [Google Scholar] [CrossRef]
- Voloshyn, I.; Schumann, C.; Cabotaje, P.R.; Zamader, A.; Land, H.; Senger, M. Secondary structure changes as the potential H(2) sensing mechanism of group D [FeFe]-hydrogenases. Chem. Commun. 2024, 60, 10914–10917. [Google Scholar] [CrossRef]
- Morra, S. Fantastic [FeFe]-Hydrogenases and where to find them. Front. Microbiol. 2022, 13, 853626. [Google Scholar] [CrossRef]
- Guo, G.; Li, Z.; Chen, L.; Ling, Q.; Zan, F.; Isawi, H.; Hao, T.; Ma, J.; Wang, Z.; Chen, G.; et al. Advances in elemental sulfur-driven bioprocesses for wastewater treatment: From metabolic study to application. Water Res. 2022, 213, 118143. [Google Scholar] [CrossRef]
- Thi Phuc, D.; Bao Yen, P.; The Hai, P.; Dam Bach Lien, L.; Ha Phuong Thao, N.; Phuong, N.M. Evaluating the sulfur oxidation capability of a Rhodopseudomonas palustris strain by gene and enzyme analyses for potential applications in environmental bioremediation. VNU J. Sci. Earth Environ. Sci. 2024, 40, 1. [Google Scholar] [CrossRef]
- Pei, P.; Aslam, M.; Wang, H.; Ye, P.; Li, T.; Liang, H.; Lin, Q.; Chen, W.; Du, H. Diversity and ecological function of urease-producing bacteria in the cultivation environment of Gracilariopsis lemaneiformis. Microb. Ecol. 2024, 87, 35. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Rani, A.; Özcan, E.; Lyu, Y.; Sela, D.A. Bifidobacterium longum subsp. infantis utilizes human milk urea to recycle nitrogen within the infant gut microbiome. Gut Microbes 2023, 15, 2192546. [Google Scholar]
- Zhou, Z.; Hashimoto, Y.; Kobayashi, M. Nitrile Degradation by Rhodococcus: Useful microbial metabolism for industrial productions. Actinomycetologica 2005, 19, 18–26. [Google Scholar] [CrossRef]
- Marron, A.O.; Akam, M.; Walker, G. Nitrile hydratase genes are present in multiple eukaryotic supergroups. PLoS ONE 2012, 7, e32867. [Google Scholar] [CrossRef]
- López-Maury, L.; Florencio, F.J.; Reyes, J.C. Arsenic sensing and resistance system in the cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 2003, 185, 5363–5371. [Google Scholar] [CrossRef]
- Wang, L.; Zhuang, X.; Zhuang, G.; Jing, C. Arsenic resistance strategy in Pantoea sp. IMH: Organization, function and evolution of ars genes. Sci. Rep. 2016, 6, 39195. [Google Scholar] [CrossRef]
- Ruffner, L.A.; Piña, M.; Beuning, P.J.; Ondrechen, M.J. Making functional predictions using local spatial arrangements in the haloacid dehalogenase superfamily. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Huang, H.; Patskovsky, Y.; Toro, R.; Farelli, J.D.; Pandya, C.; Almo, S.C.; Allen, K.N.; Dunaway-Mariano, D. Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry 2011, 50, 8937–8949. [Google Scholar] [CrossRef]
- Legatzki, A.; Grass, G.; Anton, A.; Rensing, C.; Nies, D.H. Interplay of the Czc system and two P-type ATPases in conferring metal resistance to Ralstonia metallidurans. J. Bacteriol. 2003, 185, 4354–4361. [Google Scholar] [CrossRef] [PubMed]
- Völlmecke, C.; Drees, S.L.; Reimann, J.; Albers, S.-V.; Lübben, M. The ATPases CopA and CopB both contribute to copper resistance of the thermoacidophilic archaeon Sulfolobus solfataricus. Microbiology 2012, 158, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Mealman, T.D.; Blackburn, N.J.; McEvoy, M.M. Metal export by CusCFBA, the periplasmic Cu (I)/Ag (I) transport system of Escherichia coli. Curr. Top. Membr. 2012, 69, 163–196. [Google Scholar] [PubMed]
- Yang, H.-C.; Rosen, B.P. New mechanisms of bacterial arsenic resistance. Biomed. J. 2016, 39, 5–13. [Google Scholar] [CrossRef]
- Fekih, I.; Zhang, C.; Li, Y.; Zhao, Y.; Alwathnani, H.; Saquib, Q.; Rensing, C.; Cervantes, C. Distribution of arsenic resistance genes in prokaryotes. Front. Microbiol. 2018, 9, 2473. [Google Scholar]
- Qin, J.; Rosen, B.P.; Zhang, Y.; Wang, G.; Franke, S.; Rensing, C. Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase. Proc. Natl. Acad. Sci. USA 2006, 103, 2075–2080. [Google Scholar] [CrossRef]
- Moncrief, M.B.C.; Maguire, M.E. Magnesium and the role of MgtC in growth of Salmonella typhimurium. Infect. Immun. 1998, 66, 3802–3809. [Google Scholar] [CrossRef]
- Degen, O.; Eitinger, T. Substrate specificity of nickel/cobalt permeases: Insights from mutants altered in transmembrane domains I and II. J. Bacteriol. 2002, 184, 3569–3577. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X.; Liu, G. Enhanced pollutants removal and high-value cell inclusions accumulation with Fe2+ in heavy oil refinery treatment system using Rhodopseudomonas and Pseudomonas. Chemosphere 2022, 294, 133520. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, G.; Li, J.; Li, X.; Zhang, J. Effects of metal ions on biomass and 5-aminolevulinic acid production in Rhodopseudomonas palustris wastewater treatment. Water Sci. Technol. 2016, 73, 382–388. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaysubun, C.; Chaichana, N.; Suwannasin, S.; Singkhamanan, K.; Yaikhan, T.; Kantachote, D.; Pomwised, R.; Wonglapsuwan, M.; Surachat, K. Genomic Insights and Comparative Analysis of Novel Rhodopseudomonas Species: A Purple Non-Sulfur Bacterium Isolated from Latex Rubber Sheet Wastewater. Life 2025, 15, 754. https://doi.org/10.3390/life15050754
Klaysubun C, Chaichana N, Suwannasin S, Singkhamanan K, Yaikhan T, Kantachote D, Pomwised R, Wonglapsuwan M, Surachat K. Genomic Insights and Comparative Analysis of Novel Rhodopseudomonas Species: A Purple Non-Sulfur Bacterium Isolated from Latex Rubber Sheet Wastewater. Life. 2025; 15(5):754. https://doi.org/10.3390/life15050754
Chicago/Turabian StyleKlaysubun, Chollachai, Nattarika Chaichana, Sirikan Suwannasin, Kamonnut Singkhamanan, Thunchanok Yaikhan, Duangporn Kantachote, Rattanaruji Pomwised, Monwadee Wonglapsuwan, and Komwit Surachat. 2025. "Genomic Insights and Comparative Analysis of Novel Rhodopseudomonas Species: A Purple Non-Sulfur Bacterium Isolated from Latex Rubber Sheet Wastewater" Life 15, no. 5: 754. https://doi.org/10.3390/life15050754
APA StyleKlaysubun, C., Chaichana, N., Suwannasin, S., Singkhamanan, K., Yaikhan, T., Kantachote, D., Pomwised, R., Wonglapsuwan, M., & Surachat, K. (2025). Genomic Insights and Comparative Analysis of Novel Rhodopseudomonas Species: A Purple Non-Sulfur Bacterium Isolated from Latex Rubber Sheet Wastewater. Life, 15(5), 754. https://doi.org/10.3390/life15050754