
Adeno-Associated Virus-Engineered Umbilical Cord-Derived Mesenchymal Stromal Cells Overexpressing Human sFlt-1 for Anti-Angiogenesis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Plasmids
2.2. Recombinant AAV Production
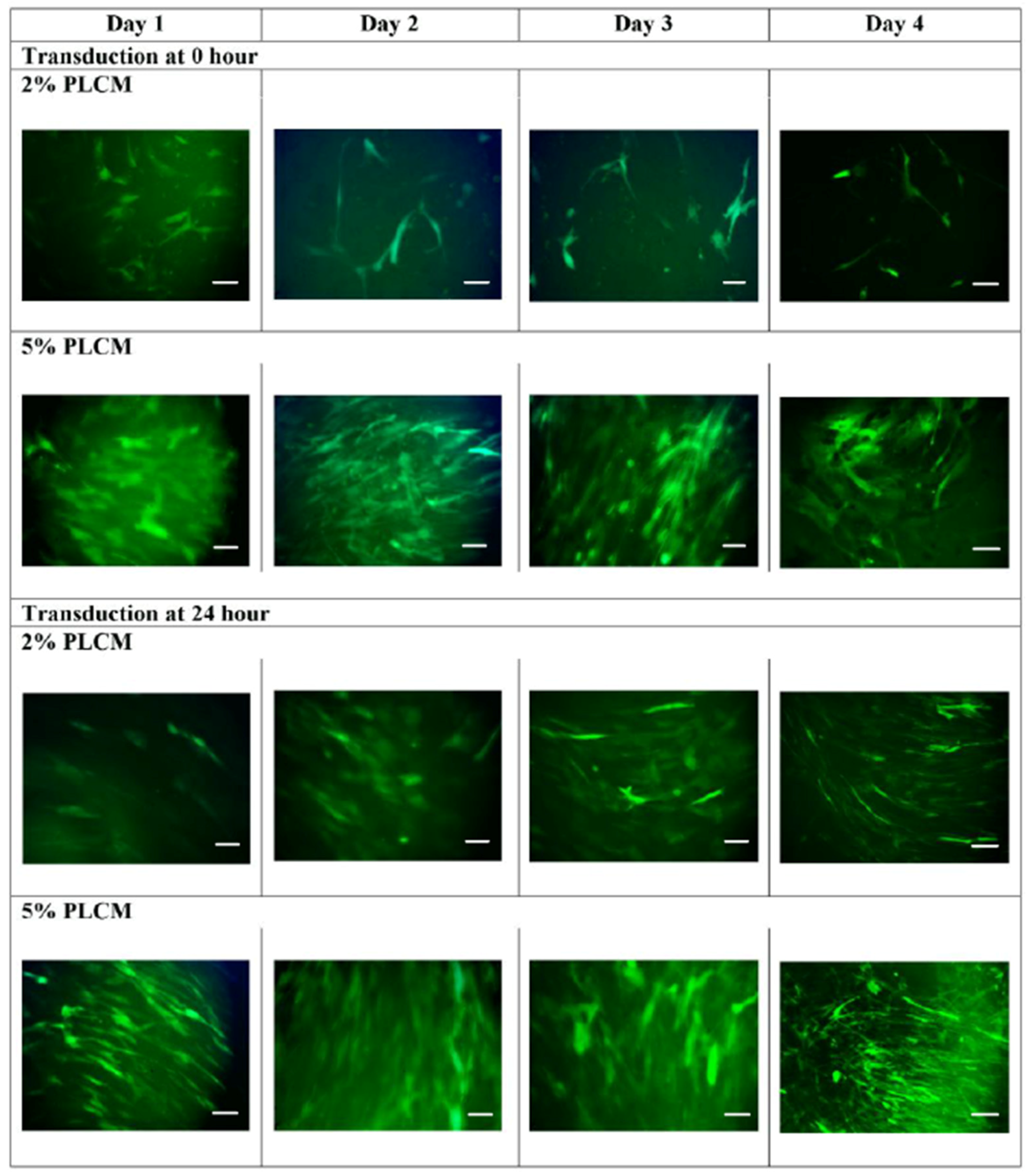
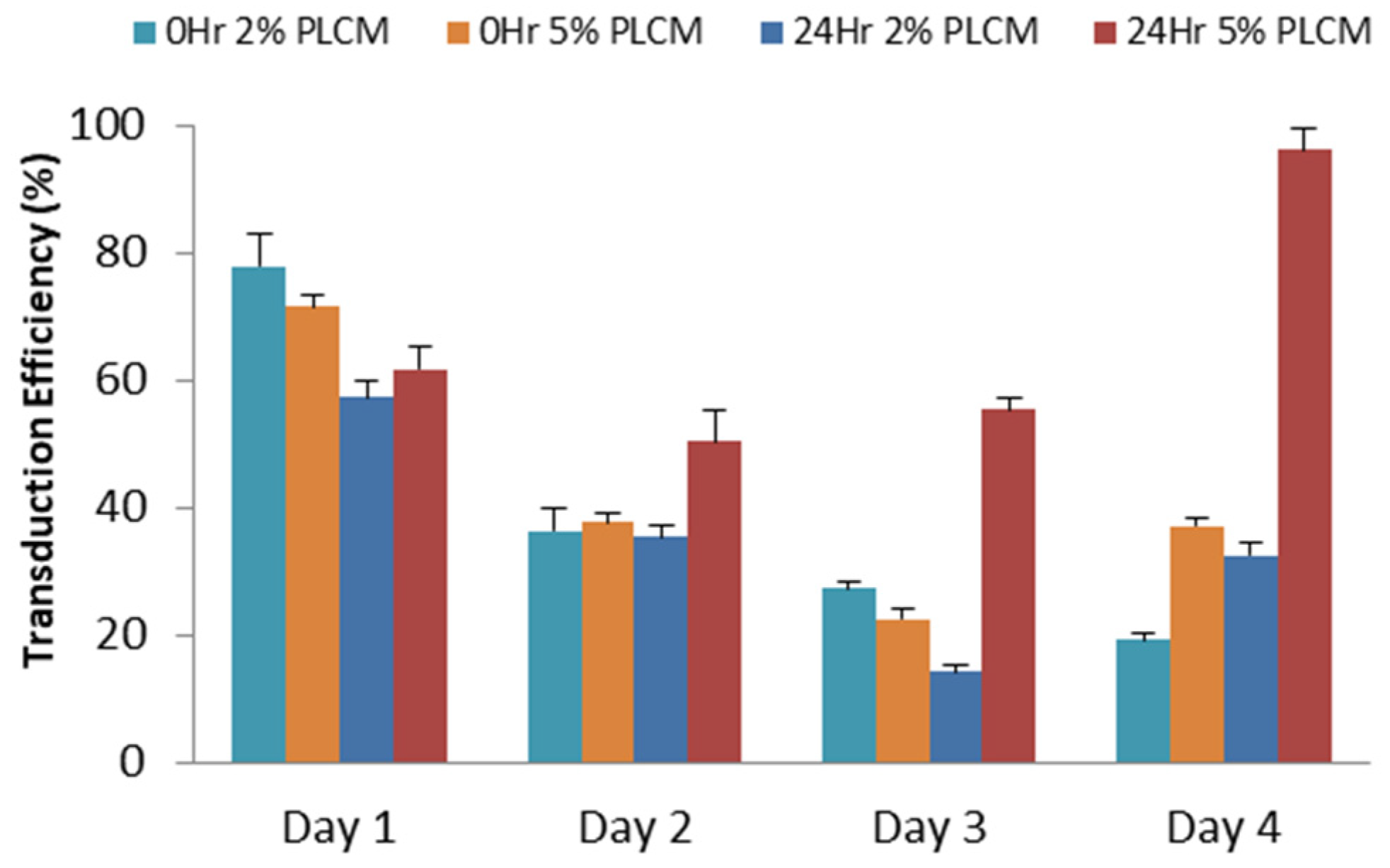
2.3. Transduction of MSCs
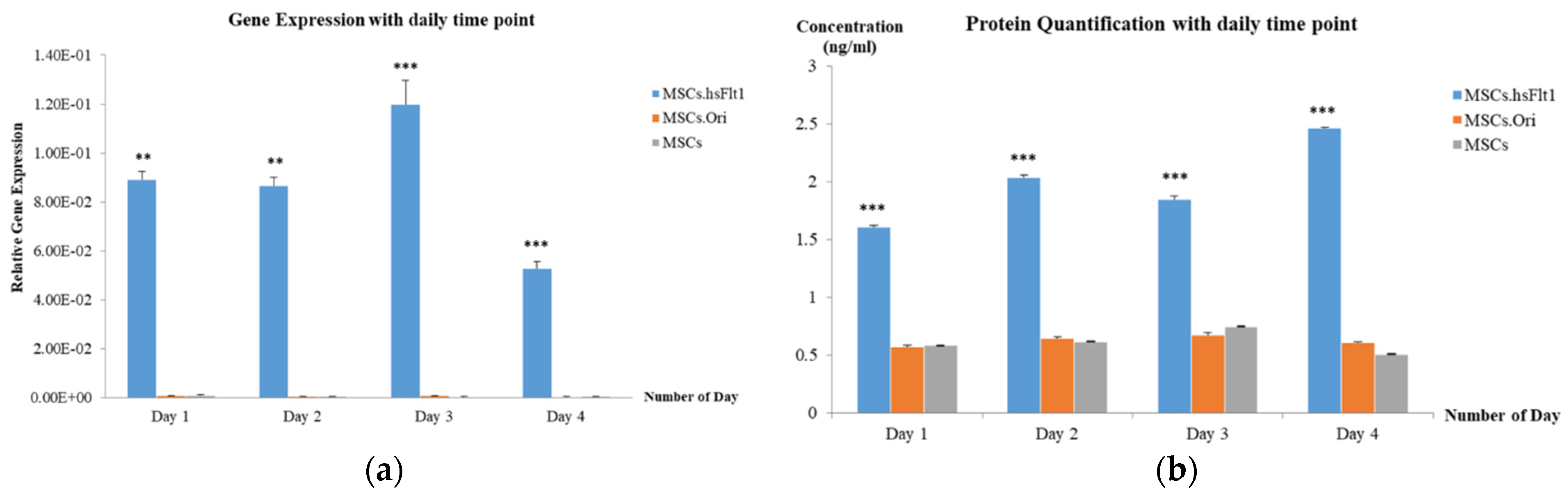
2.4. Gene Expression with qPCR
2.5. Protein Quantification with ELISA
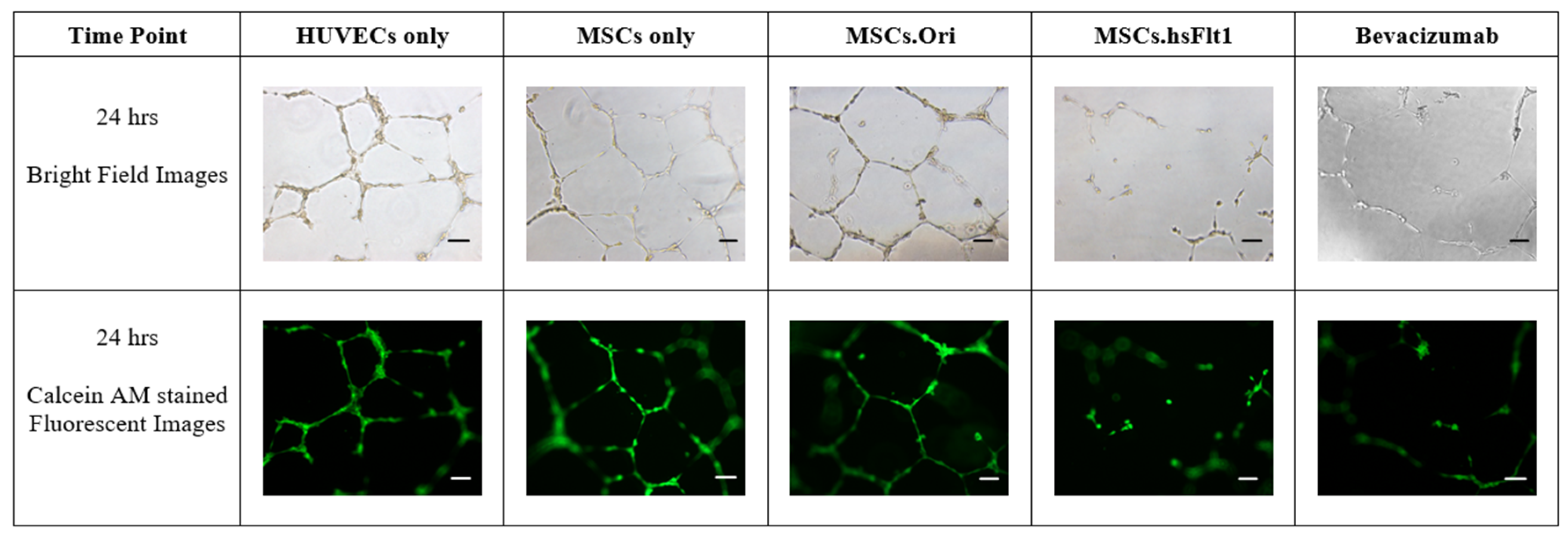
2.6. Tube Formation Assay
2.7. Statistical Analysis
3. Results
3.1. Analysis and Validation of Successful Cloning
3.2. Production of rAAV Viral Particles
3.3. Expression of hsFlt-1 in MSCs
3.4. Secretion of hsFlt-1 in MSCs
3.5. Inhibition of Tube Formation with MSCs.hsFlt1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phinney, D.G. Building a consensus regarding the nature and origin of mesenchymal stem cells. J. Cell. Biochem. 2002, 85, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Tamura, K.; Khajuria, R.K.; Bhere, D.; Nesterenko, I.; Lawler, J.; Shah, K. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol. Ther. 2015, 23, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Ewa Choy, Y.W.; Choy, K.W.; Woon, K.S.; Wafi, M.A.; Then, K.Y.; Then, K.L. Genetically Engineered Mesenchymal Stem Cells Using Viral Vectors: A New Frontier in Anti-Angiogenic Therapy. Sains Malays. 2024, 53, 63–86. [Google Scholar] [CrossRef]
- Wen, Q.; Jin, D.; Zhou, C.-Y.; Zhou, M.-Q.; Luo, W.; Ma, L. HGF-transgenic MSCs can improve the effects of tissue self-repair in a rabbit model of traumatic osteonecrosis of the femoral head. PLoS ONE 2012, 7, e37503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Z.; Ding, T.; Chen, Z.; Zhang, T. Mesenchymal stem cells overexpressing PEDF decrease the angiogenesis of gliomas. Biosci. Rep. 2013, 33, 199–205. [Google Scholar] [CrossRef]
- Obtulowicz, P.; Lech, W.; Strojek, L.; Sarnowska, A.; Domanska-Janik, K. Induction of endothelial phenotype from wharton’s jelly-derived MSCs and comparison of their vasoprotective and neuroprotective potential with primary WJ-MSCs in CA1 hippocampal region ex vivo. Cell Transplant. 2016, 25, 715–727. [Google Scholar] [CrossRef]
- Porada, C.D.; Almeida-Porada, G. Mesenchymal stem cells as therapeutics and vehicles for gene and drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 1156–1166. [Google Scholar] [CrossRef]
- Choudhery, M.S.; Badowski, M.; Muise, A.; Harris, D.T. Comparison of human mesenchymal stem cells derived from adipose and cord tissue. Cytotherapy 2013, 15, 330–343. [Google Scholar] [CrossRef]
- Guerra-Crespo, M.; Charli, J.-L.; Rosales-García, V.H.; Pedraza-Alva, G.; Pérez-Martínez, L. Polyethylenimine improves the transfection efficiency of primary cultures of post-mitotic rat fetal hypothalamic neurons. J. Neurosci. Methods 2003, 127, 179–192. [Google Scholar] [CrossRef]
- Park, H.-J.; Yang, F.; Cho, S.-W. Nonviral delivery of genetic medicine for therapeutic angiogenesis. Adv. Drug Deliv. Rev. 2012, 64, 40–52. [Google Scholar] [CrossRef]
- Basner-Tschakarjan, E.; Mingozzi, F. Cell-Mediated Immunity to AAV Vectors, Evolving Concepts and Potential Solutions. Front. Immunol. 2014, 5, 350. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yang, J.-L.; Teng, H.; Jia, Y.-Q.; Wang, R.; Zhang, X.-W.; Wu, Y.; Luo, Y.; Chen, X.-C.; Zhang, R.; et al. Anti-angiogenesis therapy based on the bone marrow-derived stromal cells genetically engineered to express sFlt-1 in mouse tumor model. BMC Cancer 2008, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Siemeister, G.; Martiny-Baron, G.; Weindel, K.; Herzog, C.; Marmé, D. Vascular endothelial growth factor up-regulates its receptor fms-like tyrosine kinase 1 (FLT-1) and a soluble variant of FLT-1 in human vascular endothelial cells. Cancer Res. 1997, 57, 5421–5425. [Google Scholar]
- Terman, B.I.; Carrion, M.; Kovacs, E.; Rasmussen, B.; Eddy, R.; Shows, T. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991, 6, 1677–1683. [Google Scholar]
- Crosson, S.M.; Dib, P.; Smith, J.K.; Zolotukhin, S. Helper-free Production of Laboratory Grade AAV and Purification by Iodixanol Density Gradient Centrifugation. Mol. Ther. Methods Clin. Dev. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.; Aminuddin, B.; Fuzina, N.; Ruszymah, B. Insulin-transferrin-selenium prevent human chondrocyte dedifferentiation and promote the formation of high quality tissue engineered human hyaline cartilage. Eur. Cells Mater. 2005, 9, 58–67. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. Allogeneic Stem Cell Transplantation: A Historical and Scientific Overview. Cancer Res. 2016, 76, 6445–6451. [Google Scholar] [CrossRef]
- Mao, F.; Xu, W.-R.; Qian, H.; Zhu, W.; Yan, Y.-M.; Shao, Q.-X.; Xu, H.-X. Immunosuppressive effects of mesenchymal stem cells in collagen-induced mouse arthritis. Inflamm. Res. 2010, 59, 219–225. [Google Scholar] [CrossRef]
- Ghannam, S.; Bouffi, C.; Djouad, F.; Jorgensen, C.; Noël, D. Immunosuppression by mesenchymal stem cells: Mechanisms and clinical applications. Stem Cell Res. Ther. 2010, 1, 2. [Google Scholar] [CrossRef]
- Nolta, J.A.; Kohn, D.B. 15—Haematopoietic stem cells for gene therapy. In Stem Cells; Potten, C.S., Ed.; Academic Press: London, UK, 1997; pp. 447–462. [Google Scholar]
- Urbán, N.; Cheung, T.H. Stem cell quiescence: The challenging path to activation. Development 2021, 148, dev165084. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, L.; Kauss, M.A.; Kopin, E.; Chandra, M.; Ul-Hasan, T.; Miller, E.; Jayandharan, G.R.; Rivers, A.E.; Aslanidi, G.V.; Ling, C.; et al. Optimizing the transduction efficiency of capsid-modified AAV6 serotype vectors in primary human hematopoietic stem cells in vitro and in a xenograft mouse model in vivo. Cytotherapy 2013, 15, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Bhukhai, K.; Yin, Z.; Tan, M.; Yoder, M.C.; Leboulch, P.; Payen, E.; Srivastava, A. High-Efficiency Transduction of Primary Human Hematopoietic Stem/Progenitor Cells by AAV6 Vectors: Strategies for Overcoming Donor-Variation and Implications in Genome Editing. Sci. Rep. 2016, 6, 35495. [Google Scholar] [CrossRef] [PubMed]
- Canté-Barrett, K.; Mendes, R.D.; Smits, W.K.; van Helsdingen-van Wijk, Y.M.; Pieters, R.; Meijerink, J.P.P. Lentiviral gene transfer into human and murine hematopoietic stem cells: Size matters. BMC Res. Notes 2016, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Ricks, D.M.; Kutner, R.; Zhang, X.-Y.; Welsh, D.A.; Reiser, J. Optimized Lentiviral Transduction of Mouse Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2008, 17, 441–450. [Google Scholar] [CrossRef]
- Lin, P.; Correa, D.; Lin, Y.; Caplan, A.I. Polybrene Inhibits Human Mesenchymal Stem Cell Proliferation during Lentiviral Transduction. PLoS ONE 2011, 6, e23891. [Google Scholar] [CrossRef]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J.; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar] [CrossRef]
- Pal, R.; Hanwate, M.; Jan, M.; Totey, S. Phenotypic and functional comparison of optimum culture conditions for upscaling of bone marrow-derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2009, 3, 163–174. [Google Scholar] [CrossRef]
- Potier, E.; Ferreira, E.; Meunier, A.; Sedel, L.; Logeart-Avramoglou, D.; Petite, H. Prolonged hypoxia concomitant with serum deprivation induces massive human mesenchymal stem cell death. Tissue Eng. 2007, 13, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S.H. Adeno-Associated Virus (AAV) Transduction of Primary Human CD4+T Lymphocytes. Master’s Thesis, Wright State University, Dayton, OH, USA, 2014. [Google Scholar]
- McMahon, J.M.; Conroy, S.; Lyons, M.; Greiser, U.; O’Shea, C.; Strappe, P.; Howard, L.; Murphy, M.; Barry, F.; O’Brien, T. Gene Transfer into Rat Mesenchymal Stem Cells: A Comparative Study of Viral and Nonviral Vectors. Stem Cells Dev. 2006, 15, 87–96. [Google Scholar] [CrossRef]
- Asuri, P.; Bartel, M.A.; Vazin, T.; Jang, J.-H.; Wong, T.B.; Schaffer, D.V. Directed Evolution of Adeno-associated Virus for Enhanced Gene Delivery and Gene Targeting in Human Pluripotent Stem Cells. Mol. Ther. 2012, 20, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Liao, W.-M.; Yuan, Z.-H.; Sheng, P.-Y.; Zhang, L.-J.; Yuan, X.-W.; Lei, L. In vitro and in vivo induction of bone formation based on adeno-associated virus-mediated BMP-7 gene therapy using human adipose-derived mesenchymal stem cells. Acta Pharmacol. Sin. 2007, 28, 839–849. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, W.I.; Heo, H.; Shin, O.; Kwon, Y.K.; Lee, H. Stable gene expression by self-complementary adeno-associated viruses in human MSCs. Biochem. Biophys. Res. Commun. 2007, 360, 573–579. [Google Scholar] [CrossRef]
- Agirrezabala, X.; Liao, H.Y.; Schreiner, E.; Fu, J.; Ortiz-Meoz, R.F.; Schulten, K.; Green, R.; Frank, J. Structural characterization of mRNA-tRNA translocation intermediates. Proc. Natl. Acad. Sci. USA 2012, 109, 6094–6099. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Gilbert, S.F. Control of Gene Expression at the Level of Translation. In Developmental Biology, 6th ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Guyette, W.A.; Matusik, R.J.; Rosen, J.M. Prolactin-mediated transcriptional and post-transcriptional control of casein gene expression. Cell 1979, 17, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Parker, R. Mechanisms of mRNA degradation in eukaryotes. Trends Biochem. Sci. 1994, 19, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Edfors, F.; Danielsson, F.; Hallström, B.M.; Käll, L.; Lundberg, E.; Pontén, F.; Forsström, B.; Uhlén, M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 2016, 12, 883. [Google Scholar] [CrossRef] [PubMed]
- Zernii, E.Y.; Baksheeva, V.E.; Iomdina, E.N.; Averina, O.A.; Permyakov, S.E.; Philippov, P.P.; Zamyatnin, A.A.; Senin, I.I. Rabbit models of ocular diseases: New relevance for classical approaches. CNS Neurol. Disord.-Drug Targets 2016, 15, 267–291. [Google Scholar] [CrossRef]
- Del Amo, E.M.; Urtti, A. Rabbit as an animal model for intravitreal pharmacokinetics: Clinical predictability and quality of the published data. Exp. Eye Res. 2015, 137, 111–124. [Google Scholar] [CrossRef]
- Gupta, S.; Martin, L.M.; Zhang, E.; Sinha, P.R.; Landreneau, J.; Sinha, N.R.; Hesemann, N.P.; Mohan, R.R. Toxicological effects of ocular acrolein exposure to eyelids in rabbits in vivo. Exp. Eye Res. 2023, 234, 109575. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence 5′–3′ |
|---|---|
| Glyceraldehyde 3-phosphate dehydrogenase (GADPH) | F: 5′-TCC CTG AGC TGA ACG GGA AG-3′ R: 5′-GGA GGA GTG GGT GTC GTC GCT GT-3′ |
| Human Fms-related tyrosine kinase 1 (hFLT1(s7)) | F: 5′-CCA TCA GCA GTT CCA CCA CT-3′ R: 5′-ACA CAG AGC CCT TCT GGT TG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choy, E.Y.-W.; Leong, C.-O.; Cheong, S.-K.; Then, K.-L.; Then, K.-Y. Adeno-Associated Virus-Engineered Umbilical Cord-Derived Mesenchymal Stromal Cells Overexpressing Human sFlt-1 for Anti-Angiogenesis. Life 2025, 15, 728. https://doi.org/10.3390/life15050728
Choy EY-W, Leong C-O, Cheong S-K, Then K-L, Then K-Y. Adeno-Associated Virus-Engineered Umbilical Cord-Derived Mesenchymal Stromal Cells Overexpressing Human sFlt-1 for Anti-Angiogenesis. Life. 2025; 15(5):728. https://doi.org/10.3390/life15050728
Chicago/Turabian StyleChoy, Ewa Yee-Wa, Chee-Onn Leong, Soon-Keng Cheong, Khong-Lek Then, and Kong-Yong Then. 2025. "Adeno-Associated Virus-Engineered Umbilical Cord-Derived Mesenchymal Stromal Cells Overexpressing Human sFlt-1 for Anti-Angiogenesis" Life 15, no. 5: 728. https://doi.org/10.3390/life15050728
APA StyleChoy, E. Y.-W., Leong, C.-O., Cheong, S.-K., Then, K.-L., & Then, K.-Y. (2025). Adeno-Associated Virus-Engineered Umbilical Cord-Derived Mesenchymal Stromal Cells Overexpressing Human sFlt-1 for Anti-Angiogenesis. Life, 15(5), 728. https://doi.org/10.3390/life15050728