


Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease

 , , , ,
, , , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Population
- No past or present medical history of autoimmune diseases, malignancies, or inflammatory chronic diseases;
- Patients with a history of kidney transplantation and/or with acute kidney injury prior to hospital admission;
- Patients receiving nephrotoxic medications before hospitalization that could confound AKI development;
- Patients with insufficient clinical or laboratory data to assess renal function at admission and during hospitalization.
2.2. Patient Stratification
- An increase in serum creatinine (SCr) by ≥0.3 mg/dL within 48 h;
- An increase in SCr to ≥1.5 times baseline within seven days;
- A urine output reduction to <0.5 mL/kg/h for more than six hours.
2.3. Data Collection and Clinical Parameters
2.4. Statistical Analysis
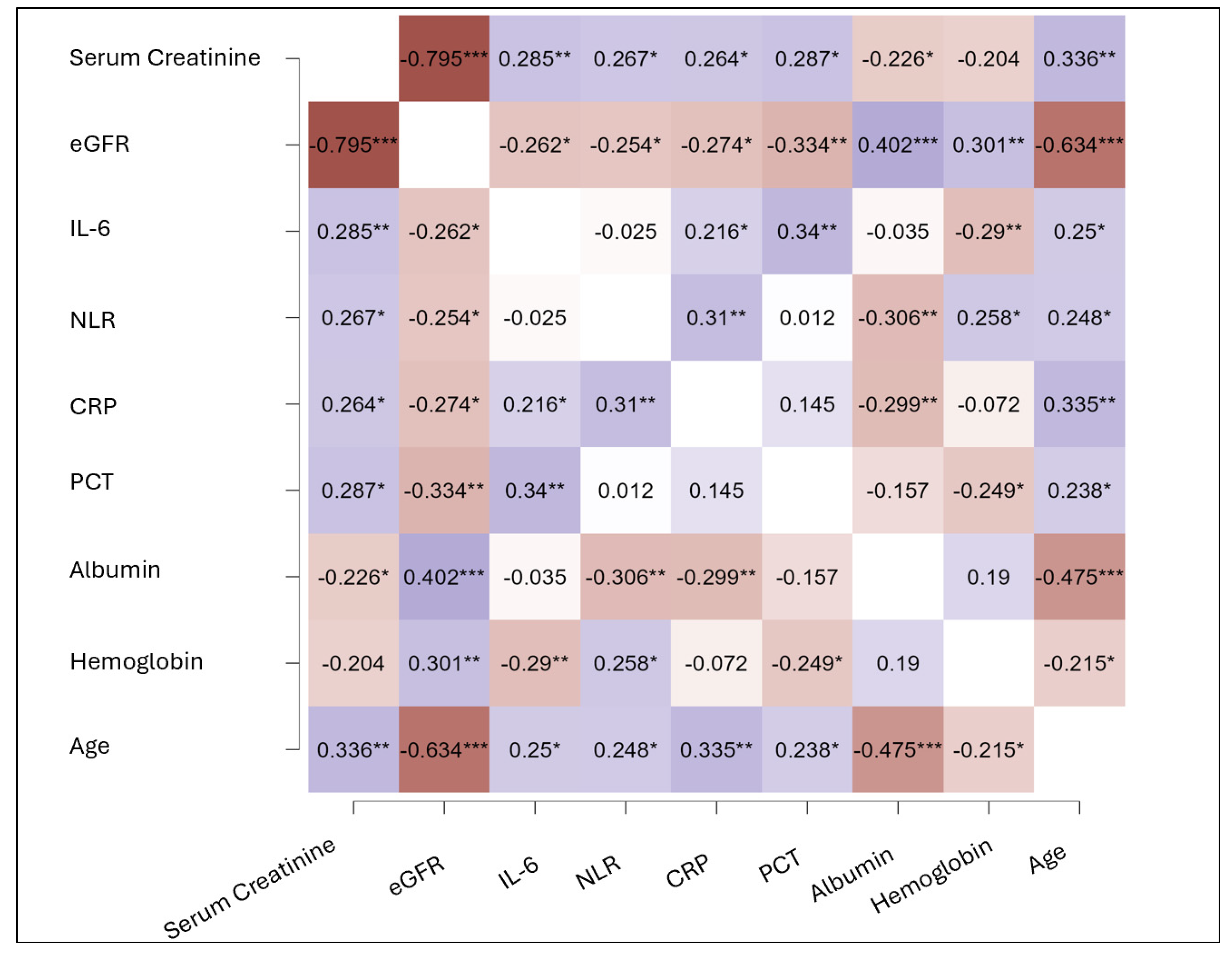
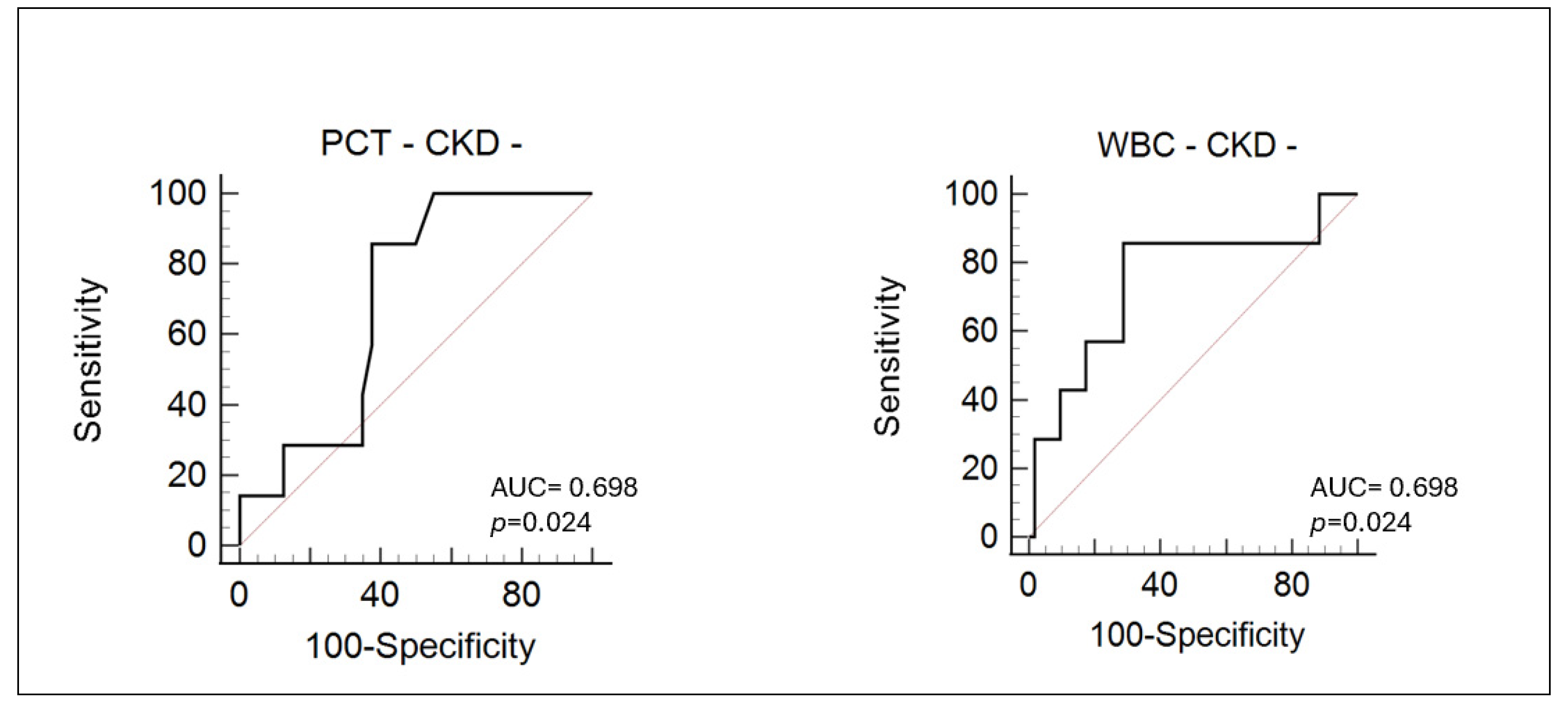
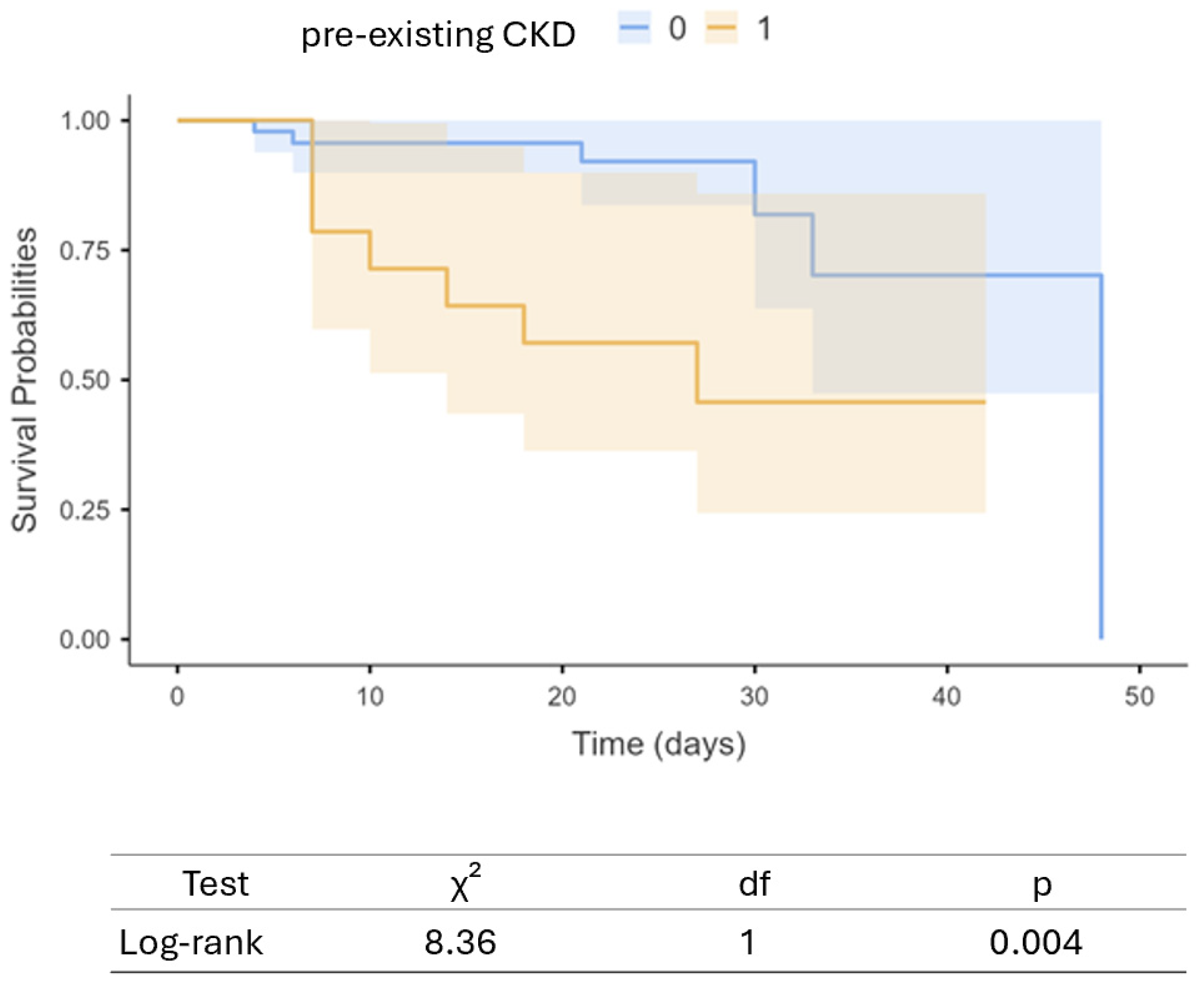
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Available online: https://www.who.int/europe/emergencies/situations/covid-19 (accessed on 5 April 2025).
- Legrand, M.; Bell, S.; Forni, L.; Joannidis, M.; Koyner, J.L.; Liu, K.; Cantaluppi, V. Pathophysiology of COVID-19-associated acute kidney injury. Nat. Rev. Nephrol. 2021, 17, 751–764. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, I.W.; Chang, L.C.; Ho, C.N.; Wu, J.-Y.; Tsai, Y.-W.; Lin, C.-M.; Chang, Y.-J.; Hung, K.-C. Association between COVID-19 and the development of chronic kidney disease in patients without initial acute kidney injury. Sci. Rep. 2025, 15, 10924. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.L.; Janse, R.J.; de Jong, Y.; van der Endt, V.H.W.; Milders, J.; van der Willik, E.M.; de Rooij, E.N.M.; Dekkers, O.M.; Rotmans, J.I.; van Diepen, M. Acute kidney injury and kidney replacement therapy in COVID-19: A systematic review and meta-analysis. Clin. Kidney J. 2020, 13, 550–563. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fabrizi, F.; Nardelli, L.; Regalia, A.; Zanoni, F.; Castellano, G. Are Kidneys Affected by SARS-CoV-2 Infection? An Updated Review on COVID-19-Associated AKI. Pathogens 2024, 13, 325. [Google Scholar] [CrossRef]
- Balan, C.; Ciuhodaru, T.; Bubenek-Turconi, S.I. Kidney Injury in Critically Ill Patients with COVID-19—From Pathophysiological Mechanisms to a Personalized Therapeutic Model. J. Crit. Care Med. 2023, 9, 148–161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- See, Y.P.; Young, B.E.; Ang, L.W.; Ooi, X.Y.; Chan, C.P.; Looi, W.L.; Yeo, S.C.; Lye, D.C. Risk Factors for Development of Acute Kidney Injury in COVID-19 Patients: A Retrospective Observational Cohort Study. Nephron 2021, 145, 256–264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell. Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Martinez-Rojas, M.A.; Vega-Vega, O.; Bobadilla, N.A. Is the kidney a target of SARS-CoV-2? Am. J. Physiol. Ren. Physiol. 2020, 318, F1454–F1462. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Silva, A.V.B.D.; Campanati, J.A.G.; Barcelos, I.S.; Santos, A.C.L.; Deus, U.P.; Soares, T.J.; Amaral, L.S.B. COVID-19 and Acute Kidney Injury—Direct and Indirect Pathophysiological Mechanisms Underlying Lesion Development. An. Acad. Bras. Cienc. 2022, 94 (Suppl. S3), e20211501. [Google Scholar] [CrossRef] [PubMed]
- Colantuoni, A.; Martini, R.; Caprari, P.; Ballestri, M.; Capecchi, P.L.; Gnasso, A.; Lo Presti, R.; Marcoccia, A.; Rossi, M.; Caimi, G. COVID-19 Sepsis and Microcirculation Dysfunction. Front. Physiol. 2020, 11, 747. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iwasaki, M.; Saito, J.; Zhao, H.; Sakamoto, A.; Hirota, K.; Ma, D. Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bülow Anderberg, S.; Luther, T.; Berglund, M.; Larsson, R.; Rubertsson, S.; Lipcsey, M.; Larsson, A.; Frithiof, R.; Hultström, M. Increased levels of plasma cytokines and correlations to organ failure and 30-day mortality in critically ill COVID-19 patients. Cytokine 2021, 138, 155389. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aroca-Martínez, G.; Musso, C.G.; Avendaño-Echavez, L.; Vélez-Verbel, M.; Chartouni-Narvaez, S.; Hernandez, S.; Hinojosa-Vidal, M.A.; Espitaleta, Z.; Cadena-Bonfanti, A. Differences between COVID-19-induced acute kidney injury and chronic kidney disease patients. J. Bras. Nefrol. 2022, 44, 155–163. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, J.Y.; Lu, J.Y.; Wang, S.; Duong, K.S.; Henry, S.; Fisher, M.C.; Duong, T.Q. Long term outcomes of patients with chronic kidney disease after COVID-19 in an urban population in the Bronx. Sci. Rep. 2025, 15, 6119. [Google Scholar] [CrossRef]
- Ran, E.; Zou, Y.; Zhao, C.; Liu, K.; Yuan, J.; Yang, W.; Zhao, L.; Yang, Q.; Yang, J.; Ju, X.; et al. COVID-19 in discharged patients with diabetes and chronic kidney disease: One-year follow-up and evaluation. Front. Endocrinol. 2025, 16, 1519993. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ponti, G.; Maccaferri, M.; Ruini, C.; Tomasi, A.; Ozben, T. Biomarkers associated with COVID-19 disease progression. Crit. Rev. Clin. Lab. Sci. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Regolo, M.; Vaccaro, M.; Sorce, A.; Stancanelli, B.; Colaci, M.; Natoli, G.; Russo, M.; Alessandria, I.; Motta, M.; Santangelo, N.; et al. Neutrophil-to-Lymphocyte Ratio (NLR) Is a Promising Predictor of Mortality and Admission to Intensive Care Unit of COVID-19 Patients. J. Clin. Med. 2022, 11, 2235. [Google Scholar] [CrossRef]
- Sayan, M.; Altinisik, H.B.; Sayan, O. Neutrophil-platelet ratio as a predictor of acute kidney injury in severe COVID-19. Medicine 2024, 103, e40053. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.M.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney disease: Improving global outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar] [CrossRef]
- John, A.K.; Norbert, L.; Peter, A.; Rashad, S.B.; Emmanuel, A.B.; Stuart, L.G.; Charles, A.H.; Michael, J.; Andreas, K.; Andrew, S.L. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- Zhang, J.; Pang, Q.; Zhou, T.; Meng, J.; Dong, X.; Wang, Z.; Zhang, A. Risk factors for acute kidney injury in COVID-19 patients: An updated systematic review and meta-analysis. Ren. Fail. 2023, 45, 2170809. [Google Scholar] [CrossRef] [PubMed]
- Silver, S.A.; Beaubien-Souligny, W.; Shah, P.S.; Harel, S.; Blum, D.; Kishibe, T.; Meraz-Munoz, A.; Wald, R.; Harel, Z. The prevalence of acute kidney injury in patients hospitalized with COVID-19 infection: A systematic review and meta-analysis. Kidney Med. 2021, 3, 83–98.e1. [Google Scholar] [CrossRef] [PubMed]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Li, X.; Liu, F.; Tian, T.; Luo, J.; Yang, Y. Acute kidney injury is associated with severe infection and fatality in patients with COVID-19: A systematic review and meta-analysis of 40 studies and 24527 patients. Pharmacol. Res. 2020, 161, 105107. [Google Scholar] [CrossRef]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D. Northwell COVID-19 Research Consortium; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef]
- Kroll, M.-K.; Schloer, S.; Candan, P.; Korthals, N.; Wenzel, C.; Ihle, H.; Gilhaus, K.; Liedtke, K.R.; Schöfbänker, M.; Surmann, B.; et al. Importance of ACE2 for SARS-CoV-2 Infection of Kidney Cells. Biomolecules 2023, 13, 472. [Google Scholar] [CrossRef]
- Barros de Lima, G.; Nencioni, E.; Thimoteo, F.; Perea, C.; Pinto, R.F.A.; Sasaki, S.D. TMPRSS2 as a Key Player in Viral Pathogenesis: Influenza and Coronaviruses. Biomolecules 2025, 15, 75. [Google Scholar] [CrossRef]
- Frithiof, R.; Bergqvist, A.; Järhult, J.D.; Lipcsey, M.; Hultström, M. Presence of SARS-CoV-2 in urine is rare and not associated with acute kidney injury in critically ill COVID-19 patients. Crit. Care 2020, 24, 587. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kudose, S.; Batal, I.; Santoriello, D.; Xu, K.; Barasch, J.; Peleg, Y.; Canetta, P.; Ratner, L.E.; Marasa, M.; Gharavi, A.G.; et al. Kidney Biopsy Findings in Patients with COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cassol, C.A.; Gokden, N.; Larsen, C.P.; Bourne, T.D. Appearances Can Be Deceiving—Viral-like Inclusions in COVID-19 Negative Renal Biopsies by Electron Microscopy. Kidney360 2020, 1, 824–828. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roufosse, C.; Curtis, E.; Moran, L.; Hollinshead, M.; Cook, T.; Hanley, B.; Horsfield, C.; Neil, D. Electron microscopic investigations in COVID-19: Not all crowns are coronas. Kidney Int. 2020, 98, 505–506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pelle, M.C.; Zaffina, I.; Lucà, S.; Forte, V.; Trapanese, V.; Melina, M.; Giofrè, F.; Arturi, F. Endothelial Dysfunction in COVID-19: Potential Mechanisms and Possible Therapeutic Options. Life 2022, 12, 1605. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Elrobaa, I.H.; New, K.J. COVID-19: Pulmonary and Extra Pulmonary Manifestations. Front. Public Health 2021, 9, 711616. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sarker, M.S. Rhabdomyolysis-related acute kidney injury in COVID-19: A critical concern. World J. Virol. 2025, 14, 100160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mendes, R.d.S.; Silva, P.L.; Robba, C.; Battaglini, D.; Lopes-Pacheco, M.; Caruso-Neves, C.; Rocco, P.R.M. Advancements in understanding the mechanisms of lung–kidney crosstalk. ICMx 2024, 12, 81. [Google Scholar] [CrossRef]
- Nazerian, Y.; Ghasemi, M.; Yassaghi, Y.; Nazerian, A.; Hashemi, S.M. Role of SARS-CoV-2-induced cytokine storm in multi-organ failure: Molecular pathways and potential therapeutic options. Int. Immunopharmacol. 2022, 113 Pt B, 109428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caimi, G.; Hopps, E.; Montana, M.; Urso, C.; Carollo, C.; Canino, B.; Presti, R.L. The function of matrix metalloproteinase-9 (MMP-9) and its tissue inhibitor (TIMP-1) in several clinical conditions: Results and analysis of our survey. Clin. Hemorheol. Microcirc. 2021, 78, 401–416. [Google Scholar] [CrossRef]
- Guarneri, M.; Scola, L.; Giarratana, R.M.; Bova, M.; Carollo, C.; Vaccarino, L.; Calandra, L.; Lio, D.; Balistreri, C.R.; Cottone, S. MIF rs755622 and IL6 rs1800795 Are Implied in Genetic Susceptibility to End-Stage Renal Disease (ESRD). Genes 2022, 13, 226. [Google Scholar] [CrossRef]
- Chen, L.D.; Hu, L.; Song, Y.; Huang, Y.P.; Yang, S.J.; Yang, J.; Zhang, X.B. Role of serum IL-6 and TNF-α in coronavirus disease 2019 (COVID-19) associated renal impairment. Eur. J. Inflamm. 2022, 20, 1721727X221126117. [Google Scholar] [CrossRef] [PubMed Central]
- Filev, R.; Lyubomirova, M.; Bogov, B.; Kalinov, K.; Hristova, J.; Svinarov, D.; Rostaing, L. IL-6 and SAA-Strong Predictors for the Outcome in COVID-19 CKD Patients. Int. J. Mol. Sci. 2023, 25, 311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, J.J.; Kuo, G.; Fan, P.C.; Lee, T.H.; Yen, C.L.; Lee, C.C.; Tian, Y.C.; Chang, C.H. Neutrophil-to-lymphocyte ratio is a marker for acute kidney injury progression and mortality in critically ill populations: A population-based, multi-institutional study. J. Nephrol. 2022, 35, 911–920. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abu Alfeilat, M.; Slotki, I.; Shavit, L. Single emergency room measurement of neutrophil/lymphocyte ratio for early detection of acute kidney injury (AKI). Intern. Emerg. Med. 2018, 13, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Ulloque-Badaracco, J.R.; Ivan Salas-Tello, W.; Al-Kassab-Córdova, A.; Alarcón-Braga, E.A.; Benites-Zapata, V.A.; Maguiña, J.L.; Hernandez, A.V. Prognostic value of neutrophil-to-lymphocyte ratio in COVID-19 patients: A systematic review and meta-analysis. Int. J. Clin. Pract. 2021, 75, e14596. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Imtiaz, F.; Shafique, K.; Mirza, S.S.; Ayoob, Z.; Vart, P.; Rao, S. Neutrophil lymphocyte ratio as a measure of systemic inflammation in prevalent chronic diseases in Asian population. Int. Arch. Med. 2012, 5, 2. [Google Scholar] [CrossRef]
- Xiang, F.-F.; Zhu, J.-M.; Cao, X.-S.; Shen, B.; Zou, J.-Z.; Liu, Z.-H.; Zhang, H.; Teng, J.; Liu, H.; Ding, X.-Q. Lymphocyte depletion and subset alteration correlate to renal function in chronic kidney disease patients. Ren. Fail. 2015, 38, 7–14. [Google Scholar] [CrossRef]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef]
- El-Hag, A.; Clark, R.A. Immunosuppression by activated human neutrophils. Dependence on the myeloperoxidase system. J. Immunol. 1987, 139, 2406–2413. [Google Scholar] [CrossRef]
- Zhao, W.M.; Tao, S.M.; Liu, G.L. Neutrophil-to-lymphocyte ratio in relation to the risk of all-cause mortality and cardiovascular events in patients with chronic kidney disease: A systematic review and meta-analysis. Ren. Fail. 2020, 42, 1059–1066. [Google Scholar] [CrossRef]
- Esa, T.; Budu, B.; Mulyono, B.; Soraya, G.V.; Usman, A.N.; Intansari, U.S. Correlation of serum interleukin-6 levels and neutrophil-lymphocyte ratio in the severity of COVID-19. F1000Research 2023, 12, 1189. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chan, L.; Chaudhary, K.; Saha, A.; Chauhan, K.; Vaid, A.; Zhao, S.; Paranjpe, I.; Somani, S.; Richter, F.; Miotto, R.; et al. AKI in Hospitalized Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 151–160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.Q.; Liu, H.; Meng, Y.; Yin, H.Y.; Gao, W.Y.; Yang, X.; Xu, D.S.; Cai, X.D.; Guan, Y.; Lerman, L.O.; et al. Critical roles of cytokine storm and secondary bacterial infection in acute kidney injury development in COVID-19: A multi-center retrospective cohort study. J. Med. Virol. 2021, 93, 6641–6652. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smarz-Widelska, I.; Grywalska, E.; Morawska, I.; Forma, A.; Michalski, A.; Mertowski, S.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Korona-Glowniak, I.; Parczewski, M.; et al. Pathophysiology and Clinical Manifestations of COVID-19-Related Acute Kidney Injury-The Current State of Knowledge and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 7082. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 84) | CKD+ (n = 25) | CKD− (n = 59) | p | |
|---|---|---|---|---|
| Age (years) | 65 ± 16 | 78 ± 11 | 59 ± 14 | <0.0001 +19 [12.6–15.4]) |
| Male sex (%) | 51 | 63 | 78 | 0.126 |
| SBP (mmHg) | 134 ± 19 | 132 ± 21 | 135 ± 17 | 0.518 |
| DBP (mmHg) | 73 ± 9 | 68 ± 8 | 76 ± 9 | <0.0001 −8 [−12–4]) |
| HR (bpm) | 84 ± 14 | 78 ± 11 | 86 ± 15 | 0.014 −8 [−14–2] |
| Baseline creatinine (mg/dL) | 0.89 (0.82–1.01) | 1.46 (1.19–2.34) | 0.79 (0.68–0.92) | <0.0001 0.67 [0.3–1] |
| Baseline eGFR (mL/min/1.73mq) | 76.0 ± 30 | 35.6 ± 13 | 92.6 ± 16 | <0.0001 −57 [−71–43] |
| Baseline IL-6 (pg/mL) | 27.4 (19.08–37.87) | 49.3 (14.2–110) | 23.6 (6.1–44.1) | 0.003 −26 [9–42] |
| Baseline CRP (mg/L) | 58.0 (40.7–74.7) | 85.3 (26.16–145.7) | 48.1 (12.4–94.7) | 0.05 |
| NLR | 5.6 (4.77–6.65) | 9.0 (5.4–22) | 5.0 (2.7–8) | <0.0001 4 [2–6] |
| PCT (µg/L) | 0.10 (0.07–0.12) | 0.19 (0.08–1.2) | 0.08 (0.05–0.12) | 0.008 0.11 [0.04–0.18] |
| Ferritin (ng/mL) | 784 (620–955) | 811 (235–1455) | 767 (453–1148) | 0.995 |
| WBC (/µL) | 7654 (6893–8713) | 8440 (4960–11,305) | 7560 (5280–10,032) | 0.282 |
| Hb (g/dL) | 13.7 (12.8–14.1) | 12.6 (10.7–14) | 14 (12.7–14.9) | 0.023 −1.4 [−2–0.4] |
| PLT (/µL) | 221,500 (203,000–252,368) | 235,000 (178,250–358,750) | 221,000 (173,250–282,000) | 0.481 |
| D-dimer (ng/mL) | 800 (604–1142) | 1173 (874–1845) | 604 (370–1309) | 0.006 569 [37–1100] |
| Fibrinogen (mg/dL) | 600 ± 195 | 673 ± 222 | 568 ± 175 | 0.024 105 [41–169] |
| CK (U/L) | 71 (56–91) | 66 (35–141) | 74 (46–110) | 0.808 |
| Albumin (g/L) | 34 (33–35) | 30 (28–34) | 35 (32–38) | 0.0002 −5 [−7–3] |
| MI (%) | 7.8 | 16 | 5 | 0.09 |
| Hypertensive heart disease (%) | 5.6 | 4 | 7 | 0.623 |
| Ischemic heart disease (%) | 11.1 | 20 | 8.5 | 0.136 |
| Stroke/TIA (%) | 3.3 | 8 | 1.7 | 0.155 |
| Heart failure (%) | 7.8 | 12 | 3.4 | 0.127 |
| Arterial hypertension (%) | 48.9 | 52 | 49 | 0.811 |
| Diabetes mellitus (%) | 35.6 | 40 | 35 | 0.702 |
| COPD (%) | 4.4 | 12 | 1.7 | 0.043 |
| AKI (%) | 18 | 32 | 11 | 0.028 |
| Length of hospital stay (days) | 19.36 ± 11.43 | 18 ± 12 | 20 ± 11 | 0.676 |
| Exitus (%) | 20 | 52 | 7 | <0.0001 |
| Parenteral nutrition during hospitalization (%) | 35 | 41 | 59 | 0.09 |
| Steroids during hospitalization (%) | 97.61 | 96 | 98 | 0.526 |
| Remdesivir (%) | 44.04 | 20 | 54 | 0.004 |
| ASA (%) | 21 | 36 | 15 | 0.034 |
| ACE-i (%) | 23.81 | 28 | 22 | 0.557 |
| ARBs (%) | 26 | 28 | 25 | 0.806 |
| Thiazide diuretics (%) | 5 | 4 | 5 | 0.831 |
| Loop diuretics (%) | 31 | 68 | 15 | <0.0001 |
| Potassium-sparing diuretics (%) | 6 | 8 | 5 | 0.606 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carollo, C.; Benfante, A.; Sorce, A.; Montalbano, K.; Cirafici, E.; Calandra, L.; Geraci, G.; Mulè, G.; Scichilone, N. Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease. Life 2025, 15, 720. https://doi.org/10.3390/life15050720
Carollo C, Benfante A, Sorce A, Montalbano K, Cirafici E, Calandra L, Geraci G, Mulè G, Scichilone N. Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease. Life. 2025; 15(5):720. https://doi.org/10.3390/life15050720
Chicago/Turabian StyleCarollo, Caterina, Alida Benfante, Alessandra Sorce, Katia Montalbano, Emanuele Cirafici, Leonardo Calandra, Giulio Geraci, Giuseppe Mulè, and Nicola Scichilone. 2025. "Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease" Life 15, no. 5: 720. https://doi.org/10.3390/life15050720
APA StyleCarollo, C., Benfante, A., Sorce, A., Montalbano, K., Cirafici, E., Calandra, L., Geraci, G., Mulè, G., & Scichilone, N. (2025). Predictive Biomarkers of Acute Kidney Injury in COVID-19: Distinct Inflammatory Pathways in Patients with and Without Pre-Existing Chronic Kidney Disease. Life, 15(5), 720. https://doi.org/10.3390/life15050720