
Nanometric and Hydrophobic Green Rust Minerals upon Exposure to Amino Acids and Nickel as Prerequisites for a Primitive Chemiosmosis

 , , ,
, , ,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Context
1.1. Top-Down; From Extant Life Back to the LUCA
1.2. Bottom-Up; From Palaeogeochemistry Towards the LUCA
1.3. The Gap Between Alkaline Hydrothermal Vents and Actual Chemiosmosis
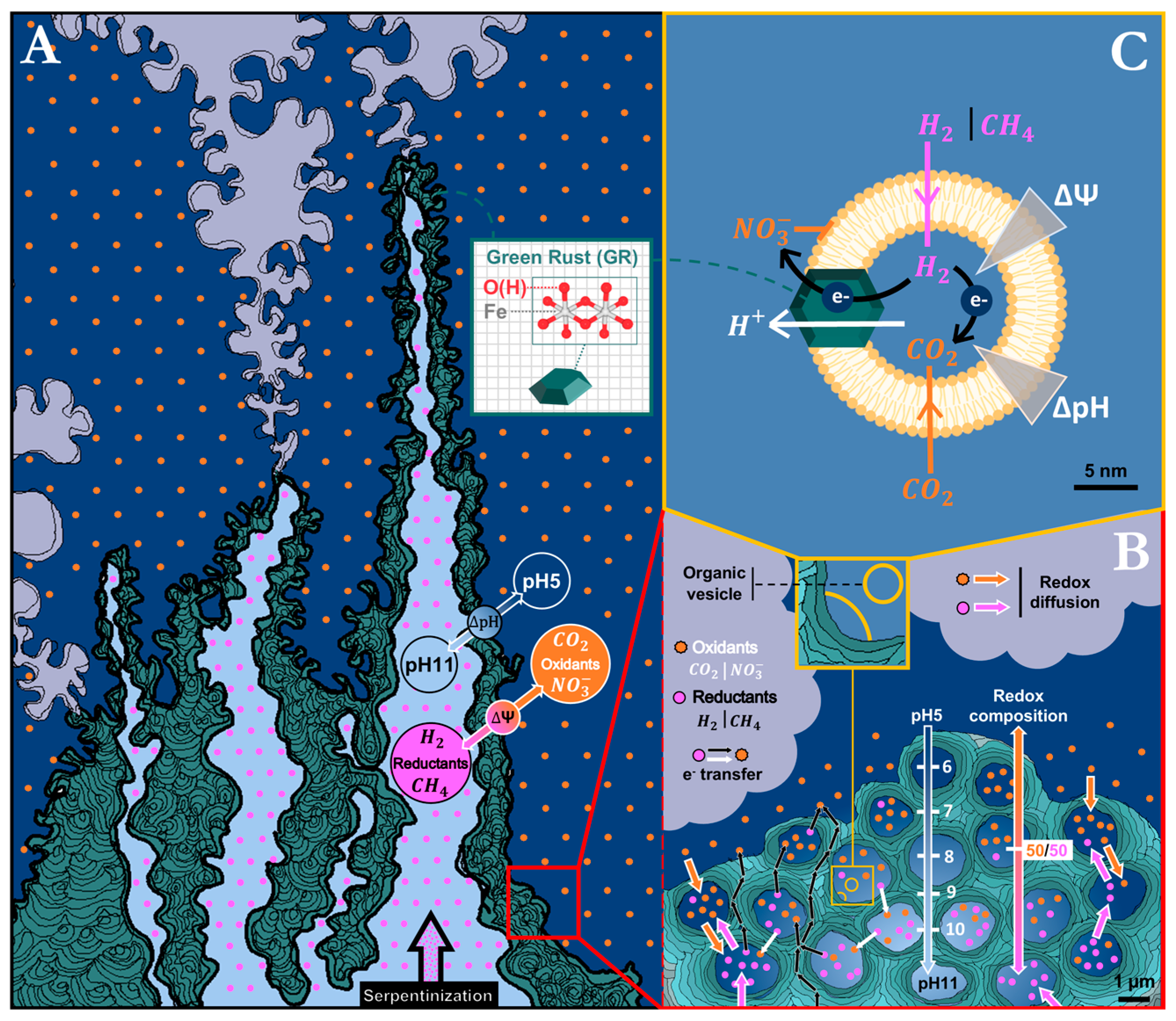
1.4. Early Organic Vesicles Coating Mineral-Walled Micropores as the Primordial Chemiosmotic Capacitors
1.5. Green Rust May Bridge the Gap
2. Introduction
3. Material and Methods
3.1. Green Rust Syntheses
3.2. Characterization of Green Rust Suspensions
3.3. Point of Zero Charge (PZC) Determination Through pH Titrations
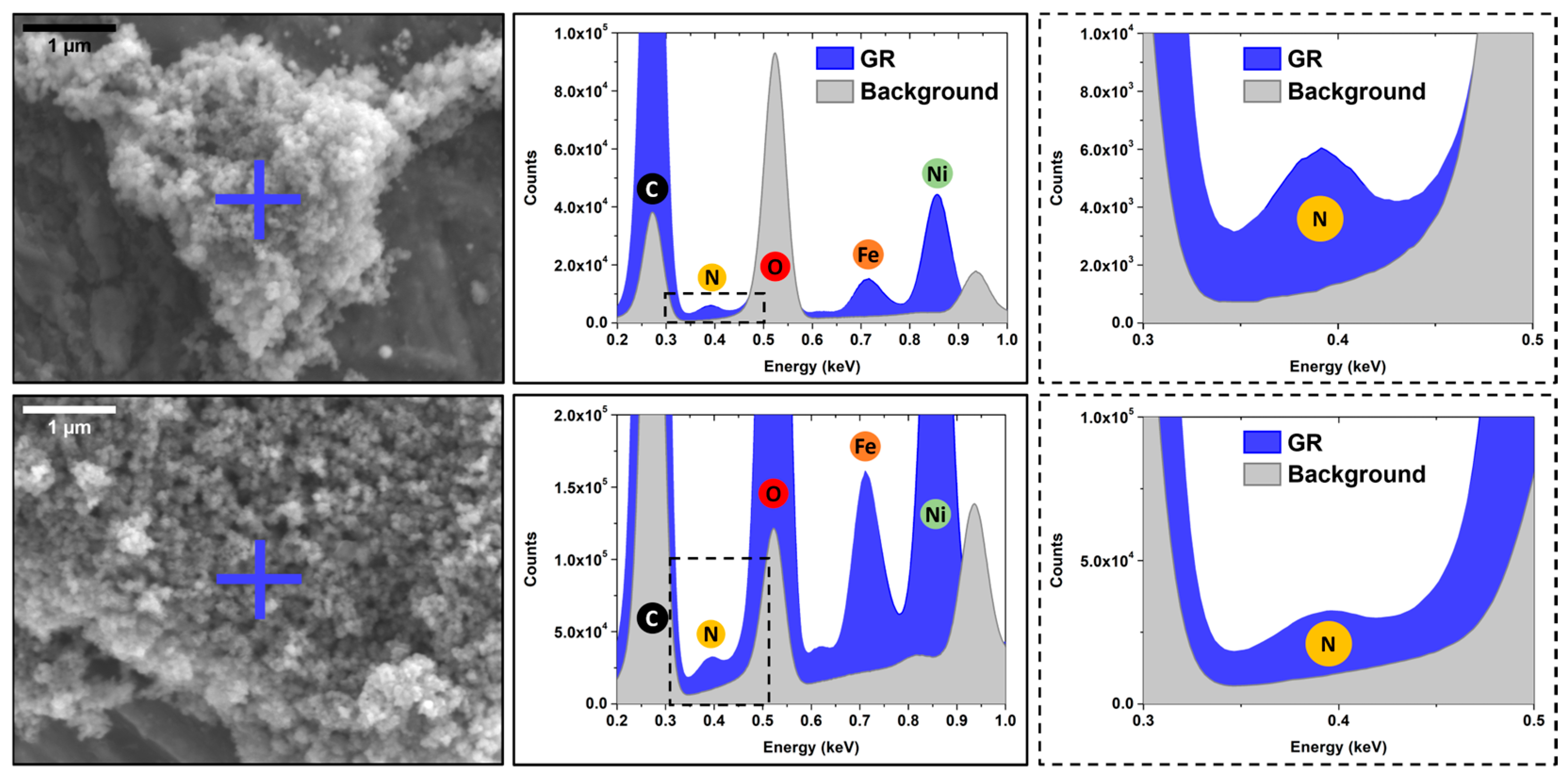
3.4. SEM-XREDS (X-Ray Energy Dispersive Spectroscopy) Chemical Analysis
3.5. ATR-FTIR Spectra Acquisition
3.6. Hydrophobicity Tests and Hydrophobic Green Rust Retrieval
3.7. Raman Spectroscopy
3.8. X-Ray Photoelectron Spectroscopy
3.9. Total Dissolved Iron Monitoring
4. Results
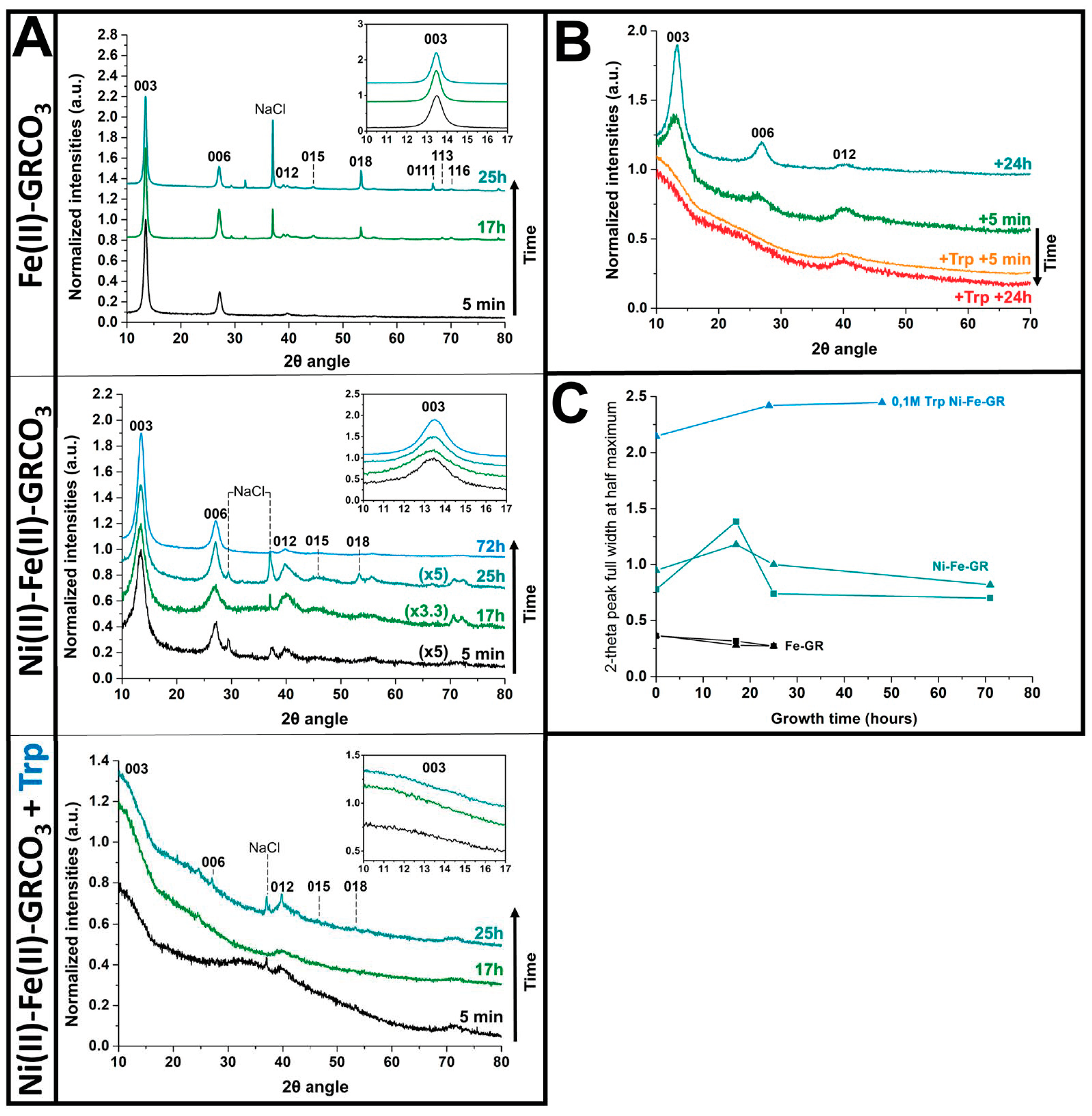
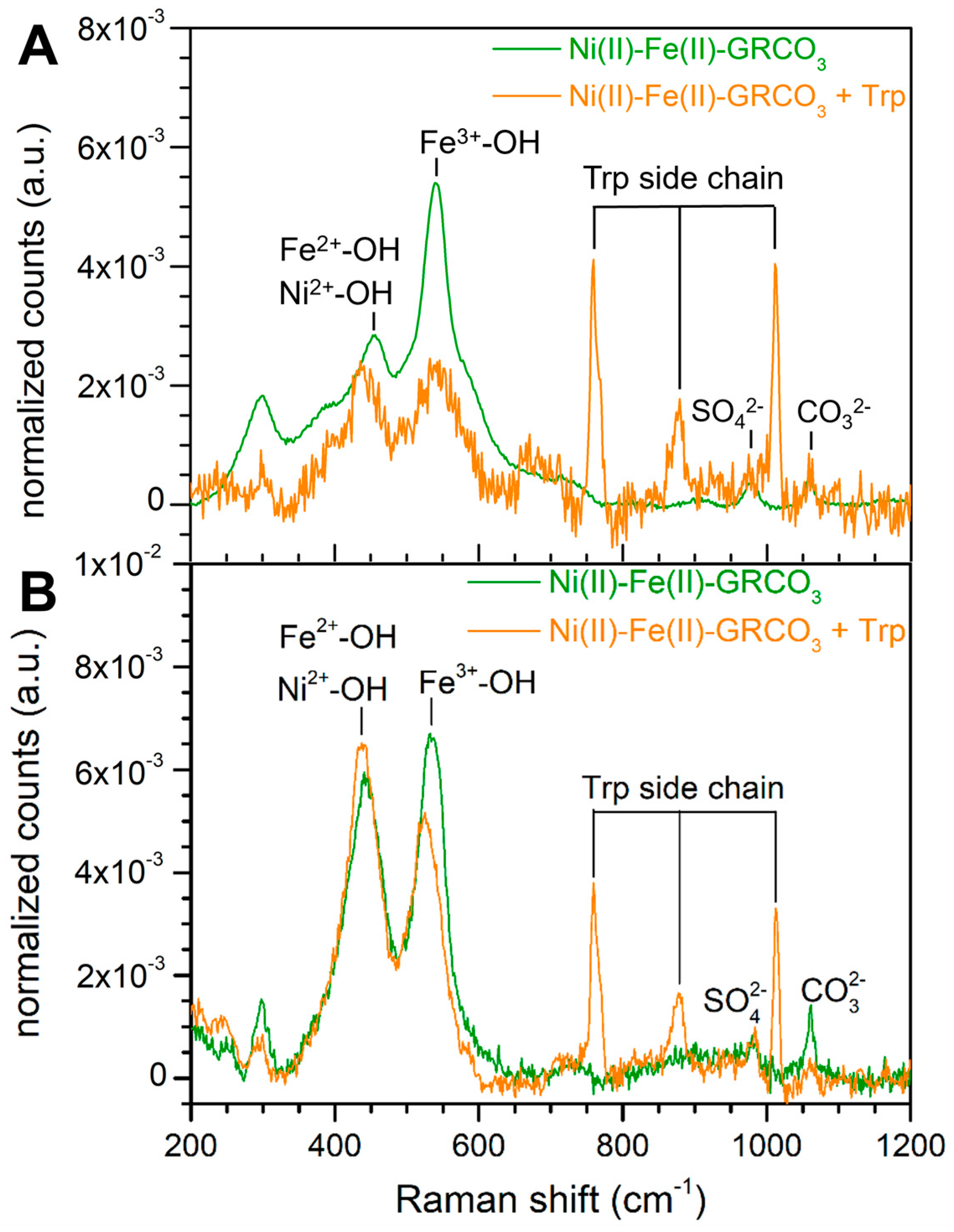
4.1. XRD and Raman Characterization of GRCO3
4.2. Electron Microscopy
4.3. Hydrophobicity Tests
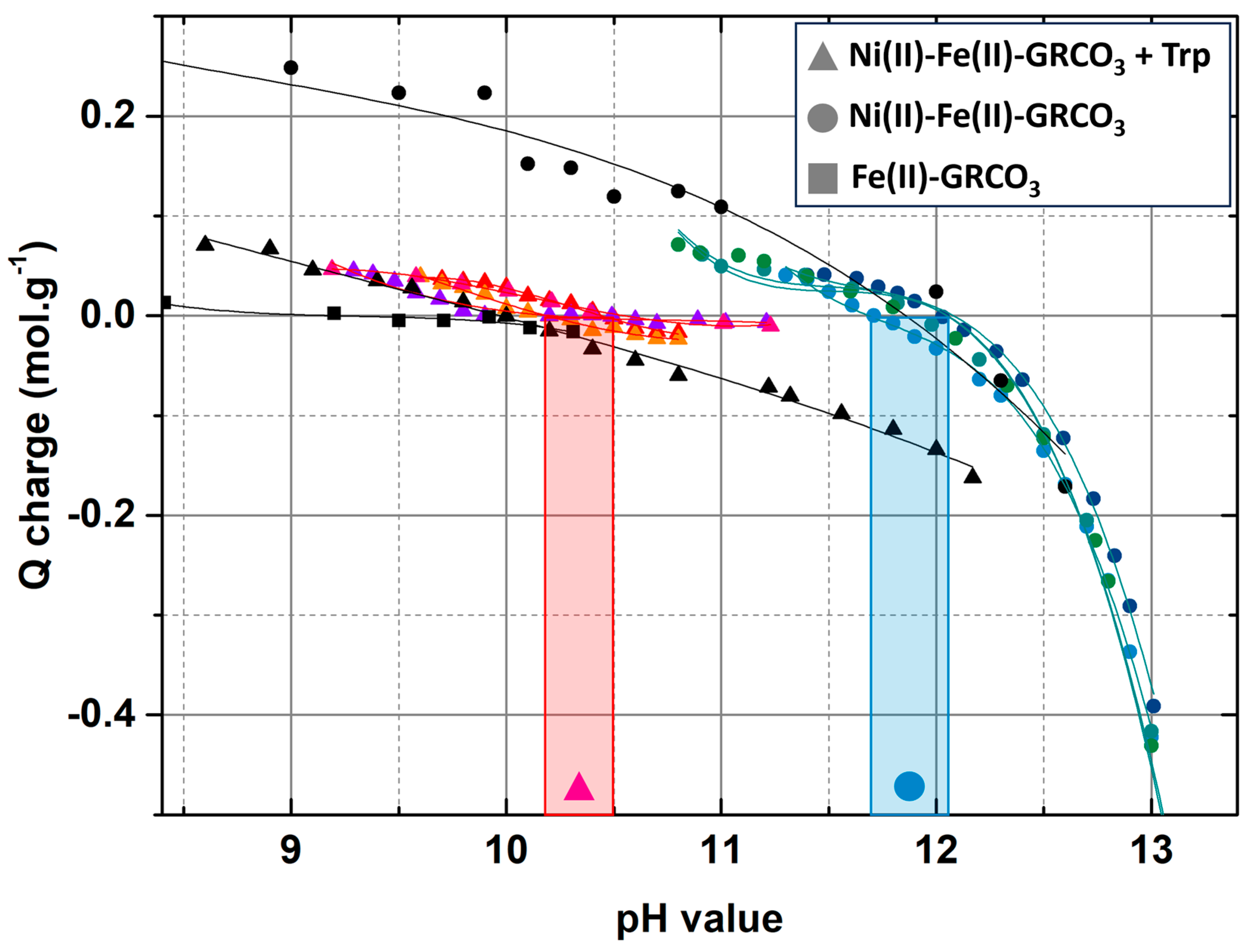
4.4. Point of Zero Charge (PZC) Determinations
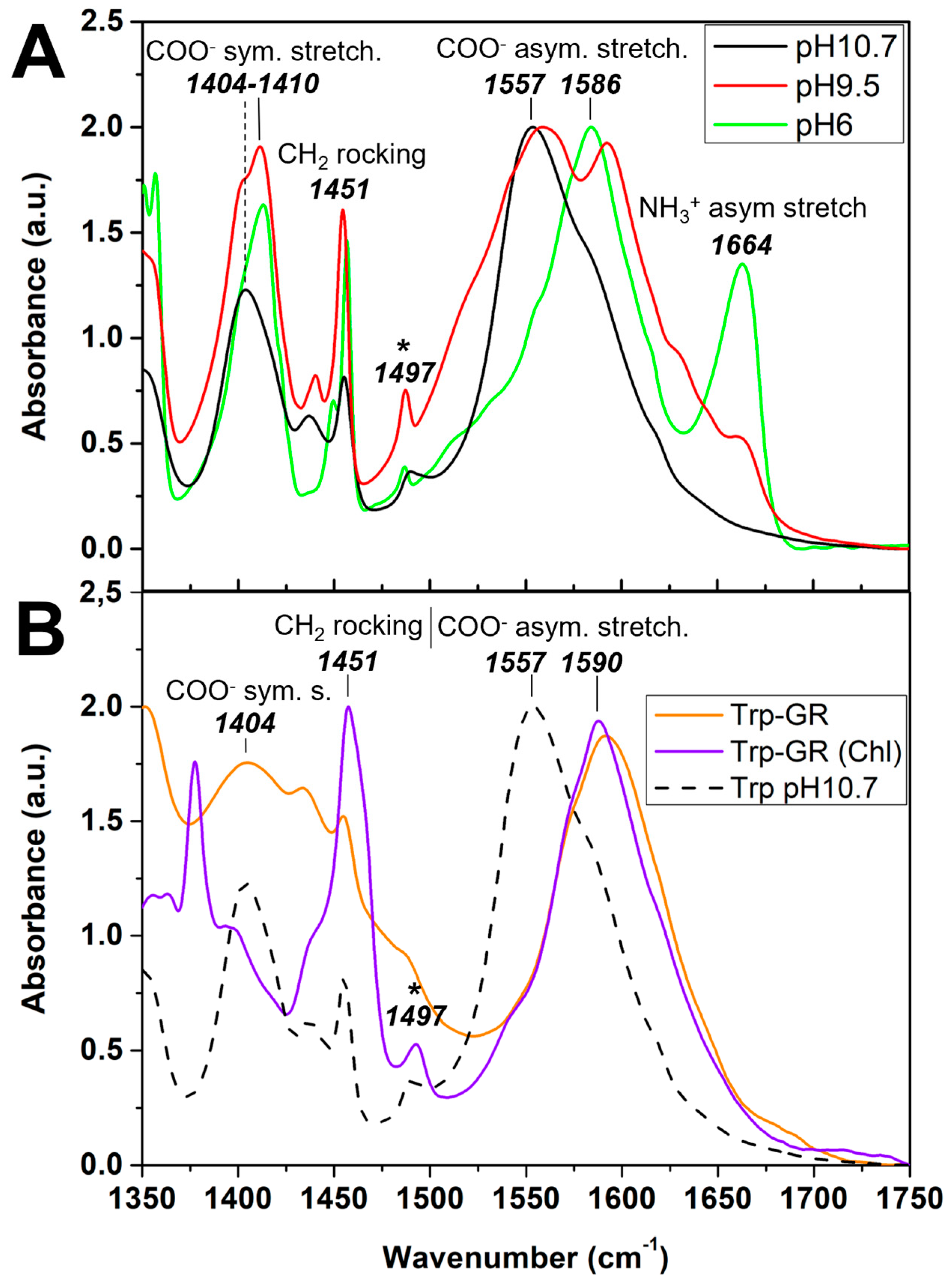
4.5. Fourier Transform-InfraRed Spectroscopy Associated to X-Ray Photoelectron Spectroscopy
5. Discussion
5.1. Ni(II) Substitution and Tryptophan Surface Adsorption Synergistically Result in Nanometric GR Crystals
5.2. Generation of Hydrophobic GR Crystals by Functionalization with Tryptophan
5.3. Implications for the Formation of AHV-Hosted Proto-Chemiosmotic Membranes
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AHV | Alkaline hydrothermal vents |
| LDH | Layered double-hydroxide |
| GR | Green rust |
| XRD | X-ray diffraction |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| PZC | Point of zero charge |
| XREDS | X-ray energy dispersive spectroscopy |
| ATR-FTIR | Attenuated total reflection-Fourier transform tnfraRed spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| Trp | Tryptophan |
| Chl | Chloroform |
References
- Schrödinger, E. What Is life? Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Needham, J. Organicism in Biology. Philosophy 1928, 3, 29–40. [Google Scholar] [CrossRef]
- Monod, J. Le Hasard et la Nécessité: Essai sur la Philosophie de la Biologie Moderne; Editions du Seuil: Paris, France, 1970. [Google Scholar]
- Prigogine, I.; Nicolis, G. Self-Organisation in Nonequilibrium Systems: Towards A Dynamics of Complexity. In Bifurcation Analysis: Principles, Applications and Synthesis; Hazewinkel, M., Jurkovich, R., Paelinck, J.H.P., Eds.; Springer: Dordrecht, The Netherlands, 1985; pp. 3–12. [Google Scholar] [CrossRef]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics: From Heat Engines to Dissipative Structures, 1st ed.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- England, J.L. Dissipative adaptation in driven self-assembly. Nat. Nanotechnol. 2015, 10, 919–923. [Google Scholar] [CrossRef]
- Endres, R.G. Entropy production selects nonequilibrium states in multistable systems. Sci. Rep. 2017, 7, 14437. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Nitschke, W.; Branscomb, E. The inevitable journey to being. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120254. [Google Scholar] [CrossRef] [PubMed]
- Branscomb, E.; Biancalani, T.; Goldenfeld, N.; Russell, M. Escapement mechanisms and the conversion of disequilibria; the engines of creation. Phys. Rep. 2017, 677, 1–60. [Google Scholar] [CrossRef]
- Lingam, M.; Loeb, A. Life in the Cosmos: From Biosignatures to Technosignatures; Harvard University Press: Cambridge, MA, USA, 2021; Available online: https://www.hup.harvard.edu/books/9780674987579 (accessed on 28 March 2025).
- Russell, M.J.; Hall, A.J.; Cairns-Smith, A.G.; Braterman, P.S. Submarine hot springs and the origin of life. Nature 1988, 336, 117. [Google Scholar] [CrossRef]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef]
- Pross, A. What Is Life? How Chemistry Becomes Biology; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Lane, N. Proton gradients at the origin of life. BioEssays 2017, 39, 1600217. [Google Scholar] [CrossRef]
- Nitschke, W.; Russell, M.J. Hydrothermal Focusing of Chemical and Chemiosmotic Energy, Supported by Delivery of Catalytic Fe, Ni, Mo/W, Co, S and Se, Forced Life to Emerge. J. Mol. Evol. 2009, 69, 481–496. [Google Scholar] [CrossRef]
- Pascal, R.; Pross, A.; Sutherland, J.D. Towards an evolutionary theory of the origin of life based on kinetics and thermodynamics. Open Biol. 2013, 3, 130156. [Google Scholar] [CrossRef]
- Sojo, V.; Herschy, B.; Whicher, A.; Camprubí, E.; Lane, N. The Origin of Life in Alkaline Hydrothermal Vents. Astrobiology 2016, 16, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Russell, M.J.; Barge, L.M.; Bhartia, R.; Bocanegra, D.; Bracher, P.J.; Branscomb, E.; Kidd, R.; McGlynn, S.; Meier, D.H.; Nitschke, W.; et al. The Drive to Life on Wet and Icy Worlds. Astrobiology 2014, 14, 308–343. [Google Scholar] [CrossRef] [PubMed]
- Schoepp-Cothenet, B.; Van Lis, R.; Atteia, A.; Baymann, F.; Capowiez, L.; Ducluzeau, A.-L.; Duval, S.; Ten Brink, F.; Russell, M.J.; Nitschke, W. On the universal core of bioenergetics. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Ferguson, S.J.; Nicholls, D.G. Bioenergetics, 4th ed.; Academic Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Nitschke, W.; McGlynn, S.E.; Milner-White, E.J.; Russell, M.J. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 871–881. [Google Scholar] [CrossRef]
- Moody, E.R.R.; Álvarez-Carretero, S.; Mahendrarajah, T.A.; Clark, J.W.; Betts, H.C.; Dombrowski, N.; Szánthó, L.L.; Boyle, R.A.; Daines, S.; Chen, X.; et al. The nature of the last universal common ancestor and its impact on the early Earth system. Nat. Ecol. Evol. 2024, 8, 1654–1666. [Google Scholar] [CrossRef]
- Nitschke, W.; Farr, O.; Gaudu, N.; Truong, C.; Guyot, F.; Russell, M.J.; Duval, S. The Winding Road from Origin to Emergence (of Life). Life 2024, 14, 607. [Google Scholar] [CrossRef]
- Kelley, D.S.; Karson, J.A.; Blackman, D.K.; Früh-Green, G.L.; Butterfield, D.A.; Lilley, M.D.; Olson, E.J.; Schrenk, M.O.; Roe, K.K.; Lebon, G.T.; et al. An off-axis hydrothermal vent field near the Mid-Atlantic Ridge at 30° N. Nature 2001, 412, 145–149. [Google Scholar] [CrossRef]
- Kelley, D.S.; Baross, J.A.; Delaney, J.R. Volcanoes, Fluids, and Life at Mid-Ocean Ridge Spreading Centers. Annu. Rev. Earth Planet. Sci. 2002, 30, 385–491. [Google Scholar] [CrossRef]
- Ludwig, K.A.; Kelley, D.S.; Butterfield, D.A.; Nelson, B.K.; Früh-Green, G. Formation and evolution of carbonate chimneys at the Lost City Hydrothermal Field. Geochim. Et Cosmochim. Acta 2006, 70, 3625–3645. [Google Scholar] [CrossRef]
- Ohara, Y.; Reagan, M.K.; Fujikura, K.; Watanabe, H.; Michibayashi, K.; Ishii, T.; Stern, R.J.; Pujana, I.; Martinez, F.; Girard, G.; et al. A serpentinite-hosted ecosystem in the Southern Mariana Forearc. Proc. Natl. Acad. Sci. USA 2012, 109, 2831–2835. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Ohara, Y.; Stern, R.J.; Yamanaka, T.; Onishi, Y.; Watanabe, H.; Chen, C.; Bloomer, S.H.; Pujana, I.; Sakai, S.; et al. Brucite chimney formation and carbonate alteration at the Shinkai Seep Field, a serpentinite-hosted vent system in the southern Mariana forearc. Geochem. Geophys. Geosyst. 2016, 17, 3775–3796. [Google Scholar] [CrossRef]
- Walker, J.C.G.; Brimblecombe, P. Iron and sulfur in the pre-biologic ocean. Precambrian Res. 1985, 28, 205–222. [Google Scholar] [CrossRef]
- White, L.M.; Bhartia, R.; Stucky, G.D.; Kanik, I.; Russell, M.J. Mackinawite and greigite in ancient alkaline hydrothermal chimneys: Identifying potential key catalysts for emergent life. Earth Planet. Sci. Lett. 2015, 430, 105–114. [Google Scholar] [CrossRef]
- Russell, M.J. Green Rust: The Simple Organizing ‘Seed’ of All Life? Life 2018, 8, 35. [Google Scholar] [CrossRef]
- Mielke, R.E.; Robinson, K.J.; White, L.M.; McGlynn, S.E.; McEachern, K.; Bhartia, R.; Kanik, I.; Russell, M.J. Iron-Sulfide-Bearing Chimneys as Potential Catalytic Energy Traps at Life’s Emergence. Astrobiology 2011, 11, 933–950. [Google Scholar] [CrossRef]
- Johnson, J.E.; Present, T.M.; Valentine, J.S. Iron: Life’s primeval transition metal. Proc. Natl. Acad. Sci. USA 2024, 121, e2318692121. [Google Scholar] [CrossRef]
- Trolard, F.; Duval, S.; Nitschke, W.; Ménez, B.; Pisapia, C.; Ben Nacib, J.; Andréani, M.; Bourrié, G. Mineralogy, geochemistry and occurrences of fougerite in a modern hydrothermal system and its implications for the origin of life. Earth-Sci. Rev. 2022, 225, 103910. [Google Scholar] [CrossRef]
- Bernal, J.D.; Dasgupta, D.R.; Mackay, A.L. The oxides and hydroxides of iron and their structural inter-relationships. Clay Miner. Bull. 1959, 4, 15–30. [Google Scholar] [CrossRef]
- Mancinelli, R.L.; McKay, C.P. The evolution of nitrogen cycling. Orig. Life Evol Biosph. 1988, 18, 311–325. [Google Scholar] [CrossRef]
- Ducluzeau, A.-L.; van Lis, R.; Duval, S.; Schoepp-Cothenet, B.; Russell, M.J.; Nitschke, W. Was nitric oxide the first deep electron sink? Trends Biochem. Sci. 2009, 34, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Charnay, B.D.; Gao, P.; Yung, Y.L.; Russell, M.J. Nitrogen Oxides in Early Earth’s Atmosphere as Electron Acceptors for Life’s Emergence. Astrobiology 2017, 17, 975–983. [Google Scholar] [CrossRef]
- Fifer, L.M.; Wong, M.L. Quantifying the Potential for Nitrate-Dependent Iron Oxidation on Early Mars: Implications for the Interpretation of Gale Crater Organics. Astrobiology 2024, 24, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.L.; Thiergart, T.; Landan, G.; Nelson-Sathi, S.; Pereira, I.A.C.; Allen, J.F.; Lane, N.; Martin, W.F. Early bioenergetic evolution. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20130088. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B. Natural pH Gradients in Hydrothermal Alkali Vents Were Unlikely to Have Played a Role in the Origin of Life. J. Mol. Evol. 2016, 83, 1–11. [Google Scholar] [CrossRef]
- Ooka, H.; McGlynn, S.E.; Nakamura, R. Electrochemistry at Deep-Sea Hydrothermal Vents: Utilization of the Thermodynamic Driving Force towards the Autotrophic Origin of Life. ChemElectroChem 2019, 6, 1316–1323. [Google Scholar] [CrossRef]
- Möller, F.M.; Kriegel, F.; Kieß, M.; Sojo, V.; Braun, D. Steep pH Gradients and Directed Colloid Transport in a Microfluidic Alkaline Hydrothermal Pore. Angew. Chem. Int. Ed. 2017, 56, 2340–2344. [Google Scholar] [CrossRef]
- Weingart, M.; Chen, S.; Donat, C.; Helmbrecht, V.; Orsi, W.D.; Braun, D.; Alim, K. Alkaline vents recreated in two dimensions to study pH gradients, precipitation morphology, and molecule accumulation. Sci. Adv. 2023, 9, eadi1884. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.J.; Ingledew, W.J. Energetic problems faced by micro-organisms growing or surviving on parsimonious energy sources and at acidic pH: I. Acidithiobacillus ferrooxidans as a paradigm. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 1471–1479. [Google Scholar] [CrossRef]
- Nakamura, R.; Takashima, T.; Kato, S.; Takai, K.; Yamamoto, M.; Hashimoto, K. Electrical Current Generation across a Black Smoker Chimney. Angew. Chem. Int. Ed. 2010, 49, 7692–7694. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nakamura, R.; Kasaya, T.; Kumagai, H.; Suzuki, K.; Takai, K. Spontaneous and Widespread Electricity Generation in Natural Deep-Sea Hydrothermal Fields. Angew. Chem. Int. Ed. 2017, 56, 5725–5728. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakamura, R.; Takai, K. Deep-Sea Hydrothermal Fields as Natural Power Plants. ChemElectroChem 2018, 5, 2162–2166. [Google Scholar] [CrossRef]
- Chakrapani, V. Unique Semiconducting Properties of Layered Mn and Fe Oxide Minerals and Their Influence on Biogeochemical Processes. In Metal Ions in Life Sciences; Taylor & Francis Group: Abingdon, UK, 2025. [Google Scholar]
- Shah, Z.H.; Riaz, S.; Atiq, S.; Naseem, S. Tunable structural and electrical impedance properties of ordered and disordered iron oxide phases for capacitive applications. Ceram. Int. 2018, 44, 16352–16364. [Google Scholar] [CrossRef]
- Wander, M.C.F.; Rosso, K.M.; Schoonen, M.A.A. Structure and Charge Hopping Dynamics in Green Rust. J. Phys. Chem. C 2007, 111, 11414–11423. [Google Scholar] [CrossRef]
- Jackson, J. Ancient Living Organisms Escaping from, or Imprisoned in, the Vents? Life 2017, 7, 36. [Google Scholar] [CrossRef]
- Sojo, V.; Ohno, A.; McGlynn, S.; Yamada, Y.; Nakamura, R. Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life 2019, 9, 16. [Google Scholar] [CrossRef]
- Hudson, R.; De Graaf, R.; Strandoo Rodin, M.; Ohno, A.; Lane, N.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R.; Barge, L.M.; Braun, D.; et al. CO2 reduction driven by a pH gradient. Proc. Natl. Acad. Sci. USA 2020, 117, 22873–22879. [Google Scholar] [CrossRef]
- Altair, T.; Dragoti, E.-S.; Sojo, V.; Li, Y.; McGlynn, S.; Galante, D.; Varela, H.; Hudson, R. Carbon reduction powered by natural electrochemical gradients under submarine hydrothermal vent conditions. chemrxiv 2024. [Google Scholar] [CrossRef]
- Hanczyc, M.M.; Mansy, S.S.; Szostak, J.W. Mineral Surface Directed Membrane Assembly. Orig. Life Evol. Biosph. 2007, 37, 67–82. [Google Scholar] [CrossRef]
- Xu, J.; Stevens, M.J.; Oleson, T.A.; Last, J.A.; Sahai, N. Role of Oxide Surface Chemistry and Phospholipid Phase on Adsorption and Self-Assembly: Isotherms and Atomic Force Microscopy. J. Phys. Chem. C 2009, 113, 2187–2196. [Google Scholar] [CrossRef]
- Nie, H.-Q.; Hou, W.-G. Vesicle formation induced by layered double hydroxides in the catanionic surfactant solution composed of sodium dodecyl sulfate and dodecyltrimethylammonium bromide. Colloid Polym. Sci. 2011, 289, 775–782. [Google Scholar] [CrossRef]
- Dalai, P.; Sahai, N. Mineral–Lipid Interactions in the Origins of Life. Trends Biochem. Sci. 2019, 44, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.F.; Rammu, H.; Zheludev, I.N.; Hartley, A.M.; Maréchal, A.; Lane, N. Promotion of protocell self-assembly from mixed amphiphiles at the origin of life. Nat. Ecol. Evol. 2019, 3, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Farr, O.; Gaudu, N.; Danger, G.; Russell, M.J.; Ferry, D.; Nitschke, W.; Duval, S. Methanol on the rocks: Green rust transformation promotes the oxidation of methane. J. R. Soc. Interface 2023, 20, 20230386. [Google Scholar] [CrossRef]
- Lee, S.; Wang, C.; Chakrapani, V. Spontaneous Unassisted Conversion of CO2 to Multicarbon Liquid Products on Green Rust Mineral. chemrxiv 2023. [Google Scholar] [CrossRef]
- Yu, F.; Fei, J.; Jia, Y.; Wang, T.; Martin, W.F.; Li, J. Chemiosmotic ATP synthesis by minimal protocells. Cell Rep. Phys. Sci. 2025, 6, 102461. [Google Scholar] [CrossRef]
- Russell, M.J. Fougerite: Free Energy Converter for Life’s Conception. In Metal Ions in Life Sciences; Taylor & Francis Group: Abingdon, UK, 2025. [Google Scholar]
- Farr, O.; Gaudu, N.; Truong, C.; Clouet, A.; Russell, M.J. Fe-oxyhydroxide “fougerite/green rust” minerals transformed (or proto-metabolized) CO2 and CH4 prior to life’s emergence. In Metal Ions in Life Sciences; Taylor & Francis Group: Abingdon, UK, 2025. [Google Scholar]
- Ruby, C.; Aïssa, R.; Géhin, A.; Cortot, J.; Abdelmoula, M.; Génin, J.-M. Green rusts synthesis by coprecipitation of FeII–FeIII ions and mass-balance diagram. Comptes Rendus Geosci. 2006, 338, 420–432. [Google Scholar] [CrossRef]
- Trolard, F.; Bourrié, G. Structure of fougerite and green rusts and a thermodynamic model for their stabilities. J. Geochem. Explor. 2006, 88, 249–251. [Google Scholar] [CrossRef]
- Ayala-Luis, K.B.; Koch, C.B.; Hansen, H.C.B. Intercalation of linear C9–C16 carboxylates in layered FeII–FeIII-hydroxides (green rust) via ion exchange. Appl. Clay Sci. 2010, 48, 334–341. [Google Scholar] [CrossRef]
- Ayala-Luis, K.B.; Koch, C.B.; Hansen, H.C.B. One-pot synthesis and characterization of FeII–FeIII hydroxide (green rust) intercalated with C9–C14 linear alkyl carboxylates. Appl. Clay Sci. 2010, 50, 512–519. [Google Scholar] [CrossRef]
- Usman, M.; Byrne, J.M.; Chaudhary, A.; Orsetti, S.; Hanna, K.; Ruby, C.; Kappler, A.; Haderlein, S.B. Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals. Chem. Rev. 2018, 118, 3251–3304. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Koch, C.B. Reduction of nitrate to ammonium by sulphate green rust: Activation energy and reaction mechanism. Clay Miner. 1998, 33, 87–101. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Guldberg, S.; Erbs, M.; Bender Koch, C. Kinetics of nitrate reduction by green rusts—Effects of interlayer anion and Fe(II):Fe(III) ratio. Appl. Clay Sci. 2001, 18, 81–91. [Google Scholar] [CrossRef]
- Choi, J.; Batchelor, B.; Won, C.; Chung, J. Nitrate reduction by green rusts modified with trace metals. Chemosphere 2012, 86, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Etique, M.; Zegeye, A.; Grégoire, B.; Carteret, C.; Ruby, C. Nitrate reduction by mixed iron(II–III) hydroxycarbonate green rust in the presence of phosphate anions: The key parameters influencing the ammonium selectivity. Water Res. 2014, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Erbs, M.; Bruun Hansen, H.C.; Olsen, C.E. Reductive Dechlorination of Carbon Tetrachloride Using Iron(II) Iron(III) Hydroxide Sulfate (Green Rust). Environ. Sci. Technol. 1999, 33, 307–311. [Google Scholar] [CrossRef]
- Ayala-Luis, K.B.; Cooper, N.G.A.; Koch, C.B.; Hansen, H.C.B. Efficient Dechlorination of Carbon Tetrachloride by Hydrophobic Green Rust Intercalated with Dodecanoate Anions. Environ. Sci. Technol. 2012, 46, 3390–3397. [Google Scholar] [CrossRef]
- Yin, W.; Strobel, B.W.; Hansen, H.C.B. Amino Acid-Assisted Dehalogenation of Carbon Tetrachloride by Green Rust: Inhibition of Chloroform Production. Environ. Sci. Technol. 2017, 51, 3445–3452. [Google Scholar] [CrossRef]
- Lee, H.-E.; Russell, M.; Nakamura, R. Water Chemistry at the Nanoscale: Clues for Resolving the “Water Paradox” Underlying the Emergence of Life. ChemistryEurope 2024, 2, e202400038. [Google Scholar] [CrossRef]
- Duval, S.; Baymann, F.; Schoepp-Cothenet, B.; Trolard, F.; Bourrié, G.; Grauby, O.; Branscomb, E.; Russell, M.J.; Nitschke, W. Fougerite: The not so simple progenitor of the first cells. Interface Focus 2019, 9, 20190063. [Google Scholar] [CrossRef] [PubMed]
- Duval, S.; Branscomb, E.; Trolard, F.; Bourrié, G.; Grauby, O.; Heresanu, V.; Schoepp-Cothenet, B.; Zuchan, K.; Russell, M.J.; Nitschke, W. On the why’s and how’s of clay minerals’ importance in life’s emergence. Appl. Clay Sci. 2020, 195, 105737. [Google Scholar] [CrossRef]
- Duval, S.; Zuchan, K.; Baymann, F.; Schoepp-Cothenet, B.; Branscomb, E.; Russell, M.J.; Nitschke, W. 5 minerals and the emergence of life. In Metals, Microbes, and Minerals—The Biogeochemical Side of Life; Kroneck, P., Sosa Torres, M., Eds.; De Gruyter: Berlin, Germany, 2021; pp. 135–158. [Google Scholar]
- Ruby, C.; Abdelmoula, M.; Naille, S.; Renard, A.; Khare, V.; Ona-Nguema, G.; Morin, G.; Génin, J.-M.R. Oxidation modes and thermodynamics of FeII–III oxyhydroxycarbonate green rust: Dissolution–precipitation versus in situ deprotonation. Geochim. Et Cosmochim. Acta 2010, 74, 953–966. [Google Scholar] [CrossRef]
- Ahmed, I.A.M.; Benning, L.G.; Kakonyi, G.; Sumoondur, A.D.; Terrill, N.J.; Shaw, S. Formation of Green Rust Sulfate: A Combined in Situ Time-Resolved X-ray Scattering and Electrochemical Study. Langmuir 2010, 26, 6593–6603. [Google Scholar] [CrossRef]
- Barthélémy, K.; Naille, S.; Despas, C.; Ruby, C.; Mallet, M. Carbonated ferric green rust as a new material for efficient phosphate removal. J. Colloid. Interface Sci. 2012, 384, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, R.; White, M.L.; Poulton, S.W. Surface charge and growth of sulphate and carbonate green rust in aqueous media. Geochim. Cosmochim. Acta 2013, 108, 141–153. [Google Scholar] [CrossRef]
- Chaves, L.H.G.; Curry, J.E.; Stone, D.A.; Chorover, J. Fate of nickel ion in (II–III) hydroxysulphate green rust synthesized by precipitation and coprecipitation. Rev. Bras. Ciênc. Solo 2007, 31, 813–818. [Google Scholar] [CrossRef]
- Chaves, L.H.G.; Curry, J.E.; Stone, D.A.; Carducci, M.D.; Chorover, J. Nickel incorporation in Fe(II, III) hydroxysulfate Green Rust: Effect on crystal lattice spacing and oxidation products. Rev. Bras. Ciênc. Solo 2009, 33, 1115–1123. [Google Scholar] [CrossRef]
- Arndt, N.T. High Ni in Archean tholeiites. Tectonophysics 1991, 187, 411–419. [Google Scholar] [CrossRef]
- Hill, R.E.T. Komatiite volcanology, volcanological setting and primary geochemical properties of komatiite-associated nickel deposits. Geochem. Explor. Environ. Anal. 2001, 1, 365–381. [Google Scholar] [CrossRef]
- Kierczak, J.; Pietranik, A.; Pędziwiatr, A. Ultramafic geoecosystems as a natural source of Ni, Cr, and Co to the environment: A review. Sci. Total. Environ. 2021, 755, 142620. [Google Scholar] [CrossRef] [PubMed]
- Gaudu, N.; Farr, O.; Ona-Nguema, G.; Duval, S. Dissolved metal ions and mineral-liposome hybrid systems: Underlying interactions, synthesis, and characterization. Biochimie 2023, 215, 100–112. [Google Scholar] [CrossRef]
- Undabeytia, T.; Mishael, Y.G.; Nir, S.; Papahadjopoulos-Sternberg, B.; Rubin, B.; Morillo, E.; Maqueda, C. A Novel System for Reducing Leaching from Formulations of Anionic Herbicides: Clay-Liposomes. Environ. Sci. Technol. 2003, 37, 4475–4480. [Google Scholar] [CrossRef]
- Nir, S.; El-Nahhal, Y.; Undabeytia, T.; Rytwo, G.; Polubesova, T.; Mishael, Y.; Rabinovitz, O.; Rubin, B. Clays, Clay Minerals, and Pesticides. In Developments in Clay Science; Elsevier: Amsterdam, The Netherlands, 2013; pp. 645–662. [Google Scholar]
- Wang, C.; Leng, S.; Xu, Y.; Tian, Q.; Zhang, X.; Cao, L.; Huang, J. Preparation of Amino Functionalized Hydrophobic Zeolite and Its Adsorption Properties for Chromate and Naphthalene. Minerals 2018, 8, 145. [Google Scholar] [CrossRef]
- Yin, Z.; Dideriksen, K.; Abdelmoula, M.; Ruby, C.; Michel, F.M.; Bjerrum, M.J.; Hansen, H.C.B. Structure of single sheet iron oxides produced from surfactant interlayered green rusts. Appl. Clay Sci. 2019, 170, 86–96. [Google Scholar] [CrossRef]
- Amstad, E.; Gillich, T.; Bilecka, I.; Textor, M.; Reimhult, E. Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions Using Dispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009, 9, 4042–4048. [Google Scholar] [CrossRef]
- Chen, Y.; Bose, A.; Bothun, G.D. Controlled Release from Bilayer-Decorated Magnetoliposomes via Electromagnetic Heating. ACS Nano 2010, 4, 3215–3221. [Google Scholar] [CrossRef]
- Amstad, E.; Kohlbrecher, J.; Müller, E.; Schweizer, T.; Textor, M.; Reimhult, E. Triggered Release from Liposomes through Magnetic Actuation of Iron Oxide Nanoparticle Containing Membranes. Nano Lett. 2011, 11, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, R.; Estelrich, J.; Busquets, M.A. Liposomes Loaded with Hydrophobic Iron Oxide Nanoparticles: Suitable T2 Contrast Agents for MRI. Int. J. Mol. Sci. 2016, 17, 1209. [Google Scholar] [CrossRef]
- Rost, N.C.V.; Sen, K.; Savliwala, S.; Singh, I.; Liu, S.; Unni, M.; Raniero, L.; Rinaldi, C. Magnetic particle imaging performance of liposomes encapsulating iron oxide nanoparticles. J. Magn. Magn. Mater. 2020, 504, 166675. [Google Scholar] [CrossRef]
- De Leo, V.; Maurelli, A.M.; Giotta, L.; Catucci, L. Liposomes containing nanoparticles: Preparation and applications. Colloids Surf. B Biointerfaces 2022, 218, 112737. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Quan, X.; Li, J.; Huo, J.; Li, X.; Zhao, Z.; Li, S.; Wan, J.; Li, J.; Liu, S.; et al. Liposomes embedded with PEGylated iron oxide nanoparticles enable ferroptosis and combination therapy in cancer. Natl. Sci. Rev. 2023, 10, nwac167. [Google Scholar] [CrossRef] [PubMed]
- Hibino, T.; Jones, W. New approach to the delamination of layered double hydroxides. J. Mater. Chem. 2001, 11, 1321–1323. [Google Scholar] [CrossRef]
- Hibino, T. Delamination of Layered Double Hydroxides Containing Amino Acids. Chem. Mater. 2004, 16, 5482–5488. [Google Scholar] [CrossRef]
- Angela, S.; Ludmila, M.; Cornelia-Ioana, I.; Denisa, F.; Cristina, C.; Natalia, P.; Sabina, G.; Mateusz, M.; Joanna, K.; Adrian-Vasile, S.; et al. Aminoacid functionalised magnetite nanoparticles Fe3O4@AA (AA = Ser, Cys, Pro, Trp) as biocompatible magnetite nanoparticles with potential therapeutic applications. Sci. Rep. 2024, 14, 26228. [Google Scholar] [CrossRef]
- Barge, L.M.; Flores, E.; Baum, M.M.; VanderVelde, D.G.; Russell, M.J. Redox and pH gradients drive amino acid synthesis in iron oxyhydroxide mineral systems. Proc. Natl. Acad. Sci. USA 2019, 116, 4828–4833. [Google Scholar] [CrossRef]
- Ménez, B.; Pisapia, C.; Andreani, M.; Jamme, F.; Vanbellingen, Q.P.; Brunelle, A.; Richard, L.; Dumas, P.; Réfrégiers, M. Abiotic synthesis of amino acids in the recesses of the oceanic lithosphere. Nature 2018, 564, 59–63. [Google Scholar] [CrossRef]
- Lambert, J.-F. Adsorption and Polymerization of Amino Acids on Mineral Surfaces: A Review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef]
- Erastova, V.; Degiacomi, M.T.; Fraser, D.G.; Greenwell, H.C. Mineral surface chemistry control for origin of prebiotic peptides. Nat. Commun. 2017, 8, 2033. [Google Scholar] [CrossRef]
- Kasprzhitskii, A.; Lazorenko, G.; Nazdracheva, T.; Yavna, V. Comparative Computational Study of L-Amino Acids as Green Corrosion Inhibitors for Mild Steel. Computation 2020, 9, 1. [Google Scholar] [CrossRef]
- Halevy, I.; Alesker, M.; Schuster, E.M.; Popovitz-Biro, R.; Feldman, Y. A key role for green rust in the Precambrian oceans and the genesis of iron formations. Nat. Geosci. 2017, 10, 135–139. [Google Scholar] [CrossRef]
- Drissi, S.H.; Refait, P.; Abdelmoula, M.; Génin, J.M.R. The preparation and thermodynamic properties of Fe(II)–Fe(III) hydroxide-carbonate (green rust 1); Pourbaix diagram of iron in carbonate-containing aqueous media. Corros. Sci. 1995, 37, 2025–2041. [Google Scholar] [CrossRef]
- Riegler, T.; Lescuyer, J.-L.; Wollenberg, P.; Quirt, D.; Beaufort, D. Alteration related to uranium deposits in the kiggavik-andrew lake structural trend, nunavut, canada: New insights from petrography and clay mineralogy. Can. Mineral. 2014, 52, 27–45. [Google Scholar] [CrossRef]
- Refait, P.; Génin, J.-M.R. The mechanisms of oxidation of ferrous hydroxychloride β-Fe2(OH)3Cl in aqueous solution: The formation of akaganeite vs goethite. Corros. Sci. 1997, 39, 539–553. [Google Scholar] [CrossRef]
- Grégoire, B.; Ruby, C.; Carteret, C. Hydrolysis of mixed Ni2+–Fe3+ and Mg2+–Fe3+ solutions and mechanism of formation of layered double hydroxides. Dalton Trans. 2013, 42, 15687–15698. [Google Scholar] [CrossRef]
- Ye, Q.; Li, L.; Li, H.; Gu, X.; Han, B.; Xu, X.; Wang, F.; Li, B. Quasi-Parallel NiFe Layered Double Hydroxide Nanosheet Arrays for Large-Current-Density Oxygen Evolution Electrocatalysis. ChemSusChem 2022, 15, e202101873. [Google Scholar] [CrossRef]
- Pearson, J.F.; Slifkin, M.A. The infrared spectra of amino acids and dipeptides. Spectrochim. Acta Part A Mol. Spectrosc. 1972, 28, 2403–2417. [Google Scholar] [CrossRef]
- Roddick-Lanzilotta, A.D.; Connor, P.A.; McQuillan, A.J. An In Situ Infrared Spectroscopic Study of the Adsorption of Lysine to TiO2 from an Aqueous Solution. Langmuir 1998, 14, 6479–6484. [Google Scholar] [CrossRef]
- Wagner, C.C.; Baran, E.J. Spectroscopic and Magnetic Behaviour of the Copper (II) Complex of L-Tryptophan. Acta Farm. Bonaer. 2004, 23, 339–342. [Google Scholar]
- Nefedova, A.; Svensson, F.G.; Vanetsev, A.S.; Agback, P.; Agback, T.; Gohil, S.; Kloo, L.; Tätte, T.; Ivask, A.; Seisenbaeva, G.A.; et al. Molecular Mechanisms in Metal Oxide Nanoparticle–Tryptophan Interactions. Inorg. Chem. 2024, 63, 8556–8566. [Google Scholar] [CrossRef]
- Olsztynska, S.; Komorowska, M.; Vrielynck, L.; Dupuy, N. Vibrational Spectroscopic Study of l-Phenylalanine: Effect of pH. Appl. Spectrosc. 2001, 55, 901–907. [Google Scholar] [CrossRef]
- Olsztynska, S.; Dupuy, N.; Vrielynck, L.; Komorowska, M. Water Evaporation Analysis of L-Phenylalanine from Initial Aqueous Solutions to Powder State by Vibrational Spectroscopy. Appl. Spectrosc. 2006, 60, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Chaudhary, S.; Jena, K.C. Self-assembly of l-phenylalanine amino acid: Electrostatic induced hindrance of fibril formation. RSC Adv. 2019, 9, 12596–12605. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.R.; De Barros, R.B.; Lourenço, J.P.; Ilharco, L.M. The Infrared Spectrum of Solid l-Alanine: Influence of pH-Induced Structural Changes. J. Phys. Chem. A 2008, 112, 8280–8287. [Google Scholar] [CrossRef]
- Hernández, B.; Pflüger, F.; Nsangou, M.; Ghomi, M. Vibrational Analysis of Amino Acids and Short Peptides in Hydrated Media. IV. Amino Acids with Hydrophobic Side Chains: L-Alanine, L-Valine, and L-Isoleucine. J. Phys. Chem. B 2009, 113, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chi, Y.S. Vibrational and electronic spectroscopic characterizations of amino acid-metal complexes. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 821–825. [Google Scholar] [CrossRef]
- Begonja, S.; Rodenas, L.A.G.; Borghi, E.B.; Morando, P.J. Adsorption of cysteine on TiO2 at different pH values: Surface complexes characterization by FTIR-ATR and Langmuir isotherms analysis. Colloids Surf. A Physicochem. Eng. Asp. 2012, 403, 114–120. [Google Scholar] [CrossRef]
- Quesada-Moreno, M.M.; Avilés-Moreno, J.R.; Márquez-García, A.Á.; Partal-Ureña, F.; López González, J.J. l-Serine in aqueous solutions at different pH: Conformational preferences and vibrational spectra of cationic, anionic and zwitterionic species. J. Mol. Struct. 2013, 1046, 136–146. [Google Scholar] [CrossRef]
- Sebben, D.; Pendleton, P. Infrared spectrum analysis of the dissociated states of simple amino acids. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 132, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Ustunol, I.B.; Gonzalez-Pech, N.I.; Grassian, V.H. pH-dependent adsorption of α-amino acids, lysine, glutamic acid, serine and glycine, on TiO2 nanoparticle surfaces. J. Colloid Interface Sci. 2019, 554, 362–375. [Google Scholar] [CrossRef]
- Stagi, L.; Sini, M.; Carboni, D.; Anedda, R.; Siligardi, G.; Gianga, T.-M.; Hussain, R.; Innocenzi, P. Modulating the poly-l-lysine structure through the control of the protonation–deprotonation state of l-lysine. Sci. Rep. 2022, 12, 19719. [Google Scholar] [CrossRef]
- Perez, J.P.H.; Freeman, H.M.; Brown, A.P.; Van Genuchten, C.M.; Dideriksen, K.; S’ari, M.; Tobler, D.J.; Benning, L.G. Direct Visualization of Arsenic Binding on Green Rust Sulfate. Environ. Sci. Technol. 2020, 54, 3297–3305. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Gutiérrez-Ariza, C.; Sainz-Díaz, C.I.; Cartwright, J.H.E. The Effect of the Presence of Amino Acids on the Precipitation of Inorganic Chemical-Garden Membranes: Biomineralization at the Origin of Life. Langmuir 2022, 38, 10538–10547. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, R.; Ebina, Y.; Iyi, N.; Sasaki, T. Positively Charged Nanosheets Derived via Total Delamination of Layered Double Hydroxides. Chem. Mater. 2005, 17, 4386–4391. [Google Scholar] [CrossRef]
- Cowley, J.M. Diffraction Physics, 3rd ed.; Elsevier Science B.V: Amsterdam, The Netherlands; New York, NY, USA, 1995. [Google Scholar]
- Williams, D.B.; Carter, C.B. (Eds.) Transmission Electron Microscopy: A Textbook for Materials Science, 2nd ed.; Springer: Boston, MA, USA, 2009. [Google Scholar] [CrossRef]
- Dobson, K.D.; McQuillan, A.J. In situ infrared spectroscopic analysis of the adsorption of aliphatic carboxylic acids to TiO2, ZrO2, Al2O3, and Ta2O5 from aqueous solutions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1999, 55, 1395–1405. [Google Scholar] [CrossRef]
- Costa, D.; Savio, L.; Pradier, C.-M. Adsorption of Amino Acids and Peptides on Metal and Oxide Surfaces in Water Environment: A Synthetic and Prospective Review. J. Phys. Chem. B 2016, 120, 7039–7052. [Google Scholar] [CrossRef]
- Jia, X.; Ma, J.; Xia, F.; Xu, Y.; Gao, J.; Xu, J. Carboxylic acid-modified metal oxide catalyst for selectivity-tunable aerobic ammoxidation. Nat. Commun. 2018, 9, 933. [Google Scholar] [CrossRef]
- Lasne, J.; Laffon, C.; Parent, P. Interaction of acetone, hydroxyacetone, acetaldehyde and benzaldehyde with the surface of water ice and HNO3·3H2O ice. Phys. Chem. Chem. Phys. 2011, 14, 697–704. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Tsukanov, A.A.; Psakhie, S.G. Energy and structure of bonds in the interaction of organic anions with layered double hydroxide nanosheets: A molecular dynamics study. Sci. Rep. 2016, 6, 19986. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaudu, N.; Truong, C.; Farr, O.; Clouet, A.; Grauby, O.; Ferry, D.; Parent, P.; Laffon, C.; Ona-Nguema, G.; Guyot, F.; et al. Nanometric and Hydrophobic Green Rust Minerals upon Exposure to Amino Acids and Nickel as Prerequisites for a Primitive Chemiosmosis. Life 2025, 15, 671. https://doi.org/10.3390/life15040671
Gaudu N, Truong C, Farr O, Clouet A, Grauby O, Ferry D, Parent P, Laffon C, Ona-Nguema G, Guyot F, et al. Nanometric and Hydrophobic Green Rust Minerals upon Exposure to Amino Acids and Nickel as Prerequisites for a Primitive Chemiosmosis. Life. 2025; 15(4):671. https://doi.org/10.3390/life15040671
Chicago/Turabian StyleGaudu, Nil, Chloé Truong, Orion Farr, Adriana Clouet, Olivier Grauby, Daniel Ferry, Philippe Parent, Carine Laffon, Georges Ona-Nguema, François Guyot, and et al. 2025. "Nanometric and Hydrophobic Green Rust Minerals upon Exposure to Amino Acids and Nickel as Prerequisites for a Primitive Chemiosmosis" Life 15, no. 4: 671. https://doi.org/10.3390/life15040671
APA StyleGaudu, N., Truong, C., Farr, O., Clouet, A., Grauby, O., Ferry, D., Parent, P., Laffon, C., Ona-Nguema, G., Guyot, F., Nitschke, W., & Duval, S. (2025). Nanometric and Hydrophobic Green Rust Minerals upon Exposure to Amino Acids and Nickel as Prerequisites for a Primitive Chemiosmosis. Life, 15(4), 671. https://doi.org/10.3390/life15040671