
Comparison of Three DNA Isolation Methods and Two Sequencing Techniques for the Study of the Human Microbiota

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
1.1. Role of the Microbiome in Breast Cancer and the Importance of Microbiome Research
1.2. Sequencing Strategies and Their Significance in Microbiome Studies
1.3. Challenges in Microbiome Research and Rationale
2. Materials and Methods
2.1. Design
2.2. Biological Samples
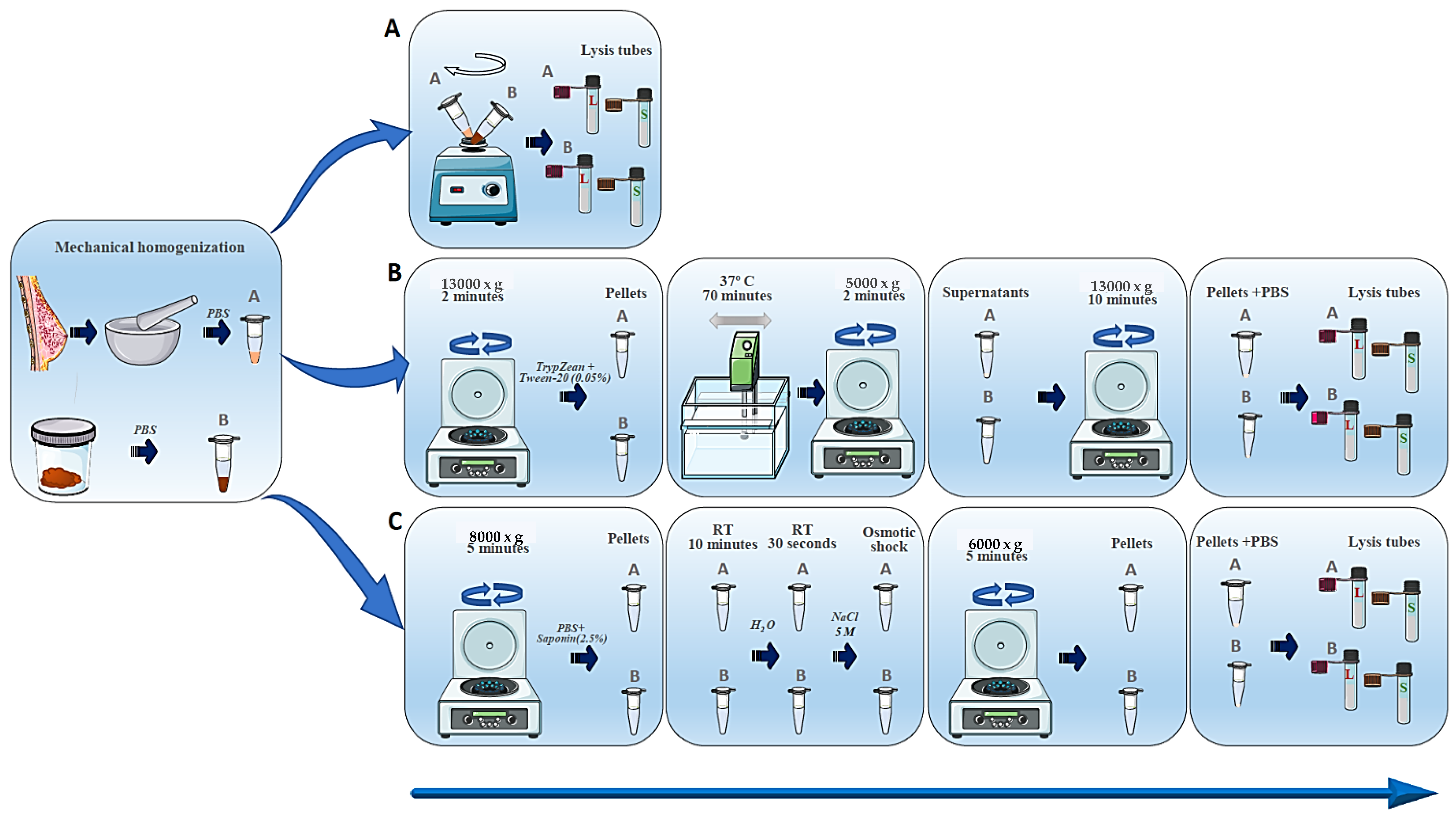
2.3. DNA Isolation Methods
2.3.1. Sample Lysis
2.3.2. DNA Extraction
2.4. DNA Sequencing
2.5. Bioinformatic Analyses
2.5.1. 16S rRNA Method
2.5.2. Shotgun Method
2.6. Statistic Analysis
3. Results
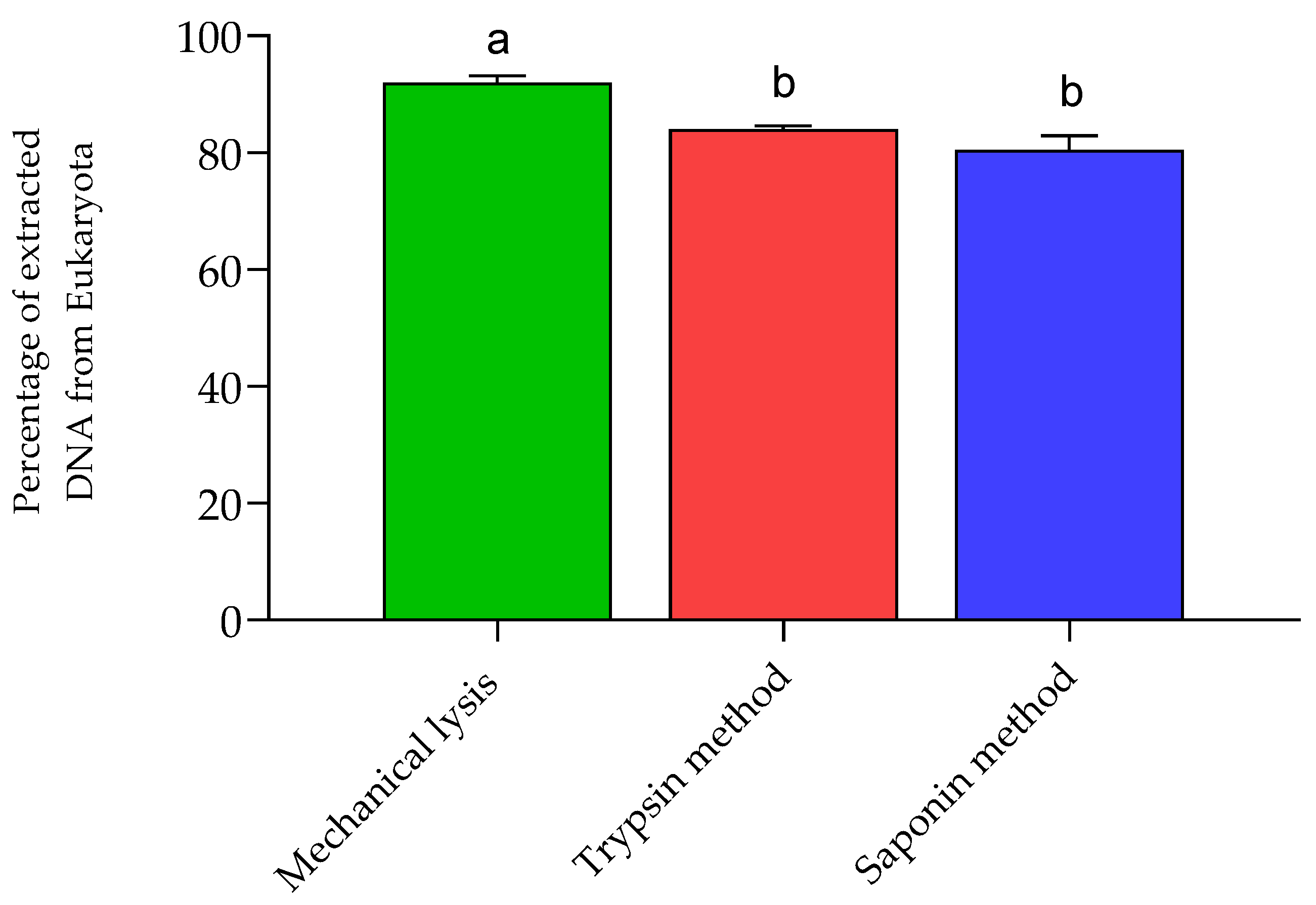
3.1. Human DNA Depletion
3.2. 16S rRNA Sequencing
3.3. Shotgun
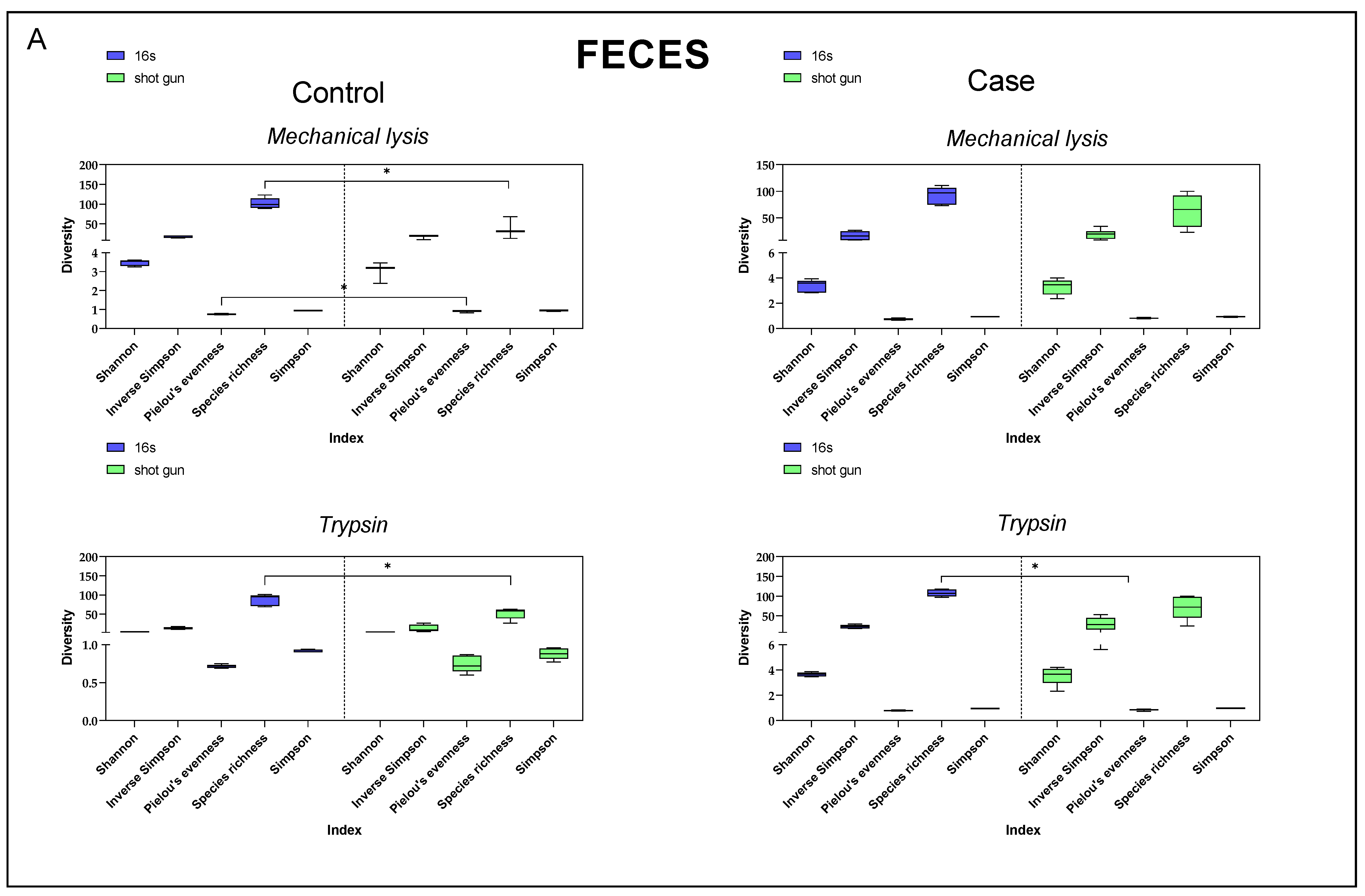
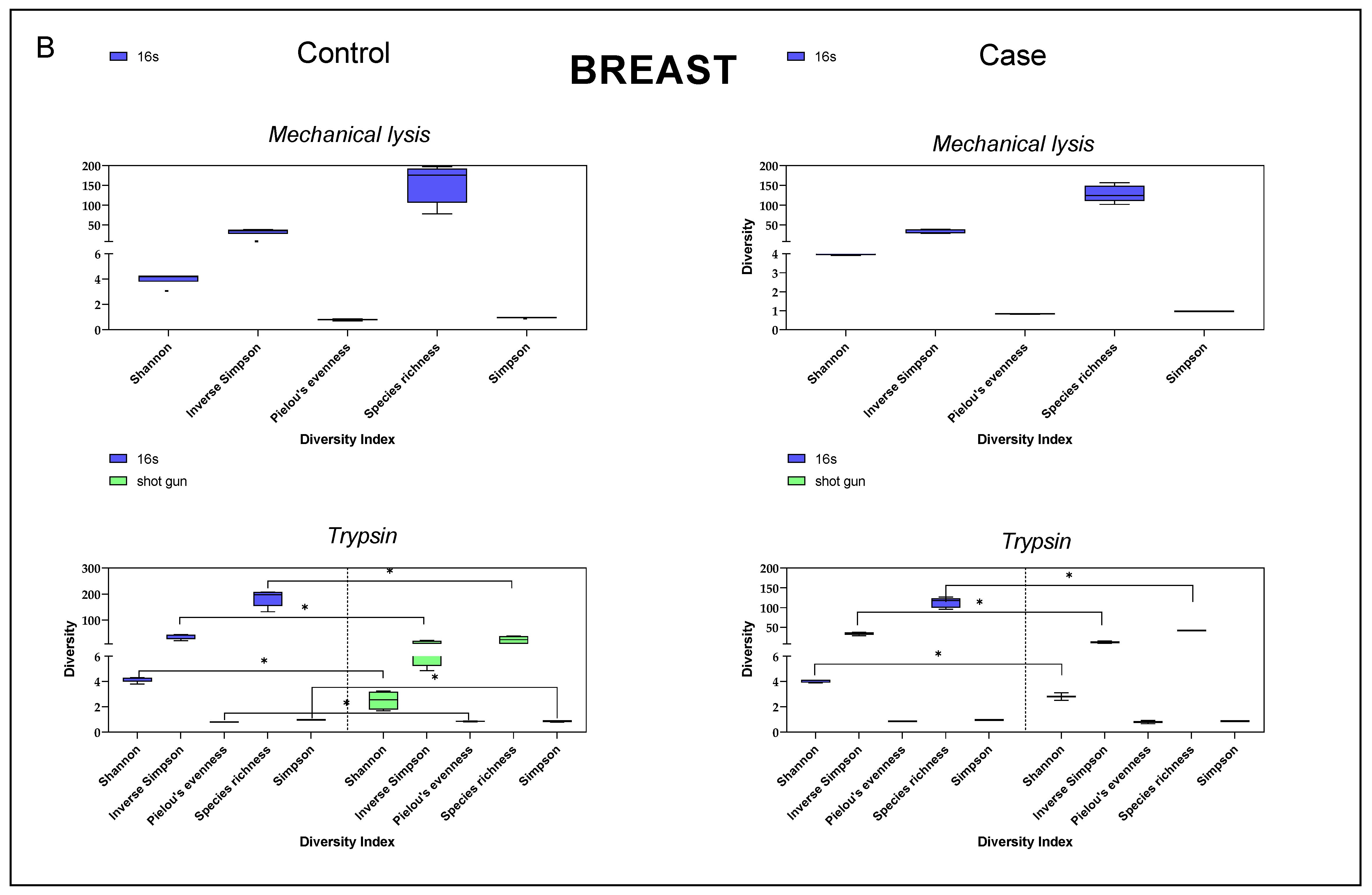
3.4. Diversity Index Comparison
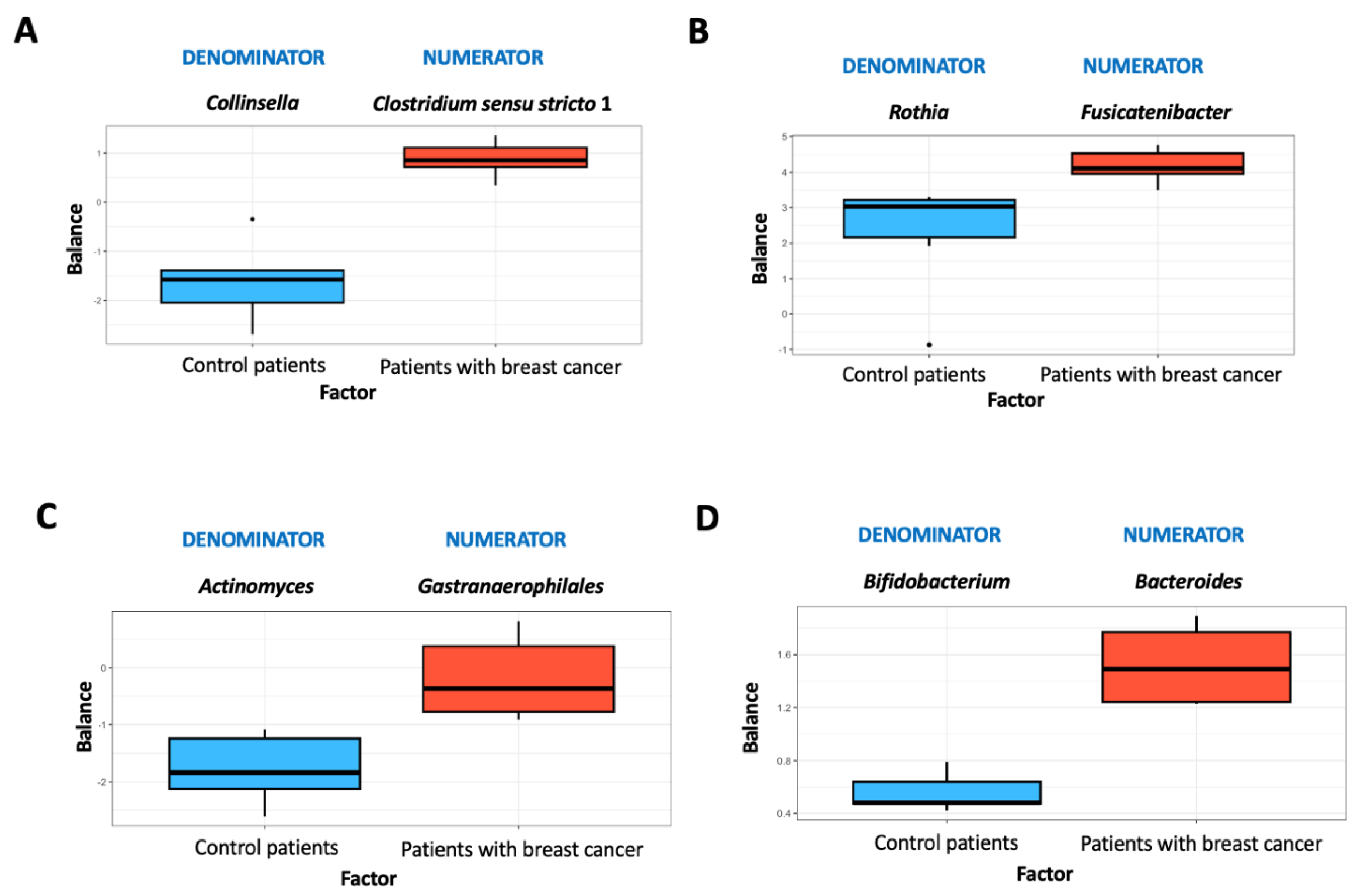
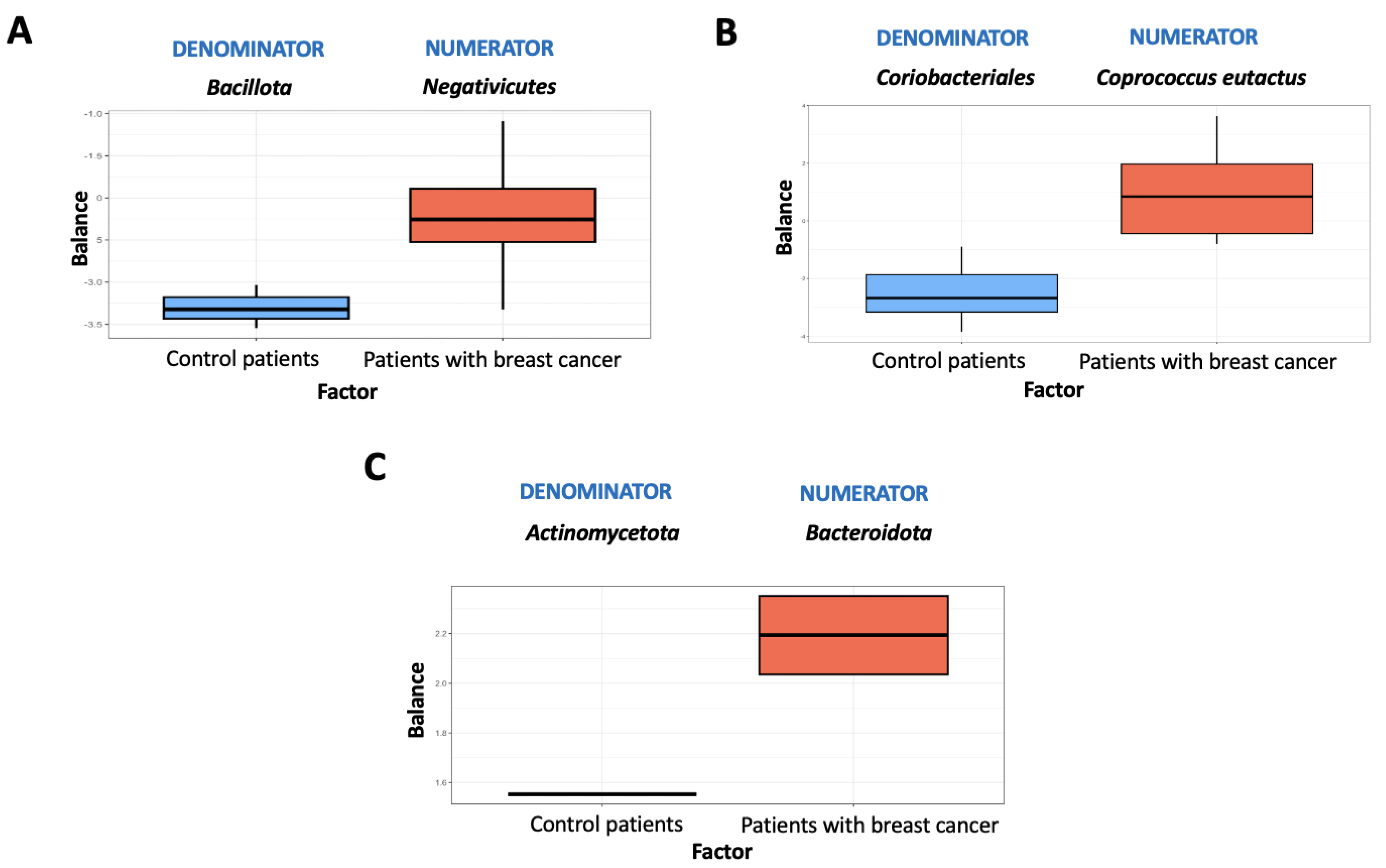
3.5. Microbiome Balance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The Human Gut Microbiota and Virome: Potential Therapeutic Implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Díaz, J.; Álvarez-Mercado, A.I.; Ruiz-Marín, C.M.; Reina-Pérez, I.; Pérez-Alonso, A.J.; Sánchez-Andujar, M.B.; Torné, P.; Gallart-Aragón, T.; Sánchez-Barrón, M.T.; Reyes Lartategui, S.; et al. Association of Breast and Gut Microbiota Dysbiosis and the Risk of Breast Cancer: A Case-Control Clinical Study. BMC Cancer 2019, 19, 495. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor–Positive Female Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef]
- Rossini, A.; Rumio, C.; Sfondrini, L.; Tagliabue, E.; Morelli, D.; Miceli, R.; Mariani, L.; Palazzo, M.; Ménard, S.; Balsari, A. Influence of Antibiotic Treatment on Breast Carcinoma Development in Proto- Neu Transgenic Mice. Cancer Res. 2006, 66, 6219–6224. [Google Scholar] [CrossRef]
- Velicer, C.M.; Lampe, J.W.; Heckbert, S.R.; Potter, J.D.; Taplin, S.H. Hypothesis: Is Antibiotic Use Associated with Breast Cancer. Cancer Causes Control 2003, 14, 739–747. [Google Scholar] [CrossRef]
- Reid, G. Can Breast Microbiota Provide Protective Effects Against Cancer? Future Microbiol. 2016, 11, 987–989. [Google Scholar] [CrossRef]
- Roderburg, C.; Loosen, S.H.; Joerdens, M.S.; Demir, M.; Luedde, T.; Kostev, K. Antibiotic Therapy Is Associated with an Increased Incidence of Cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, L.; Liu, Y.; Ren, H.; Shen, Y.; Bi, F.; Zhang, T.; Wang, X. Alter between Gut Bacteria and Blood Metabolites and the Anti-Tumor Effects of Faecalibacterium Prausnitzii in Breast Cancer. BMC Microbiol. 2020, 20, 82. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, Y.; Sun, J. Breast and Gut Microbiome in Health and Cancer. Genes Dis. 2021, 8, 581–589. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Chadha, J.; Nandi, D.; Atri, Y.; Nag, A. Significance of Human Microbiome in Breast Cancer: Tale of an Invisible and an Invincible. Semin. Cancer Biol. 2021, 70, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.K.; Gardner, S.N.; Marti, J.M.; Slezak, T.R.; Gokhale, M.B.; Allen, J.E. Using Populations of Human and Microbial Genomes for Organism Detection in Metagenomes. Genome Res. 2015, 25, 1056–1067. [Google Scholar] [CrossRef]
- Zhu, J.; Liao, M.; Yao, Z.; Liang, W.; Li, Q.; Liu, J.; Yang, H.; Ji, Y.; Wei, W.; Tan, A.; et al. Breast Cancer in Postmenopausal Women Is Associated with an Altered Gut Metagenome. Microbiome 2018, 6, 136. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Sevim, V.; Lee, J.; Egan, R.; Clum, A.; Hundley, H.; Lee, J.; Everroad, R.C.; Detweiler, A.M.; Bebout, B.M.; Pett-Ridge, J.; et al. Shotgun Metagenome Data of a Defined Mock Community Using Oxford Nanopore, PacBio and Illumina Technologies. Sci. Data 2019, 6, 285. [Google Scholar] [CrossRef]
- Nelson, M.T.; Pope, C.E.; Marsh, R.L.; Wolter, D.J.; Weiss, E.J.; Hager, K.R.; Vo, A.T.; Brittnacher, M.J.; Radey, M.C.; Hayden, H.S.; et al. Human and Extracellular DNA Depletion for Metagenomic Analysis of Complex Clinical Infection Samples Yields Optimized Viable Microbiome Profiles. Cell Rep. 2019, 26, 2227–2240.e5. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.; Hofmann, P.; Majda, S.; Dahms, E.; Dröge, J.; Fiedler, J.; Lesker, T.R.; Belmann, P.; DeMaere, M.Z.; Darling, A.E.; et al. CAMISIM: Simulating Metagenomes and Microbial Communities. Microbiome 2019, 7, 17. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Maidak, B.L. The RDP (Ribosomal Database Project) Continues. Nucleic Acids Res. 2000, 28, 173–174. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Zou, B.; Li, J.; Zhou, Q.; Quan, Z.-X. MIPE: A Metagenome-Based Community Structure Explorer and SSU Primer Evaluation Tool. PLoS ONE 2017, 12, e0174609. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S. Shotgun DNA Sequencing Using Cloned DNase I-Generated Fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef]
- Bars-Cortina, D.; Ramon, E.; Rius-Sansalvador, B.; Guinó, E.; Garcia-Serrano, A.; Mach, N.; Khannous-Lleiffe, O.; Saus, E.; Gabaldón, T.; Ibáñez-Sanz, G.; et al. Comparison between 16S rRNA and Shotgun Sequencing in Colorectal Cancer, Advanced Colorectal Lesions, and Healthy Human Gut Microbiota. BMC Genom. 2024, 25, 730. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards Standards for Human Fecal Sample Processing in Metagenomic Studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Knudsen, B.E.; Bergmark, L.; Munk, P.; Lukjancenko, O.; Priemé, A.; Aarestrup, F.M.; Pamp, S.J. Impact of Sample Type and DNA Isolation Procedure on Genomic Inference of Microbiome Composition. mSystems 2016, 1, e00095-16. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Clooney, A.G.; Stanton, C.; Claesson, M.J.; Cotter, P.D. 16S rRNA Gene Sequencing of Mock Microbial Populations- Impact of DNA Extraction Method, Primer Choice and Sequencing Platform. BMC Microbiol. 2016, 16, 123. [Google Scholar] [CrossRef]
- Albertsen, M.; Karst, S.M.; Ziegler, A.S.; Kirkegaard, R.H.; Nielsen, P.H. Back to Basics—The Influence of DNA Extraction and Primer Choice on Phylogenetic Analysis of Activated Sludge Communities. PLoS ONE 2015, 10, e0132783. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of Bacterial DNA Extraction Method from Fecal Material Influences Community Structure as Evaluated by Metagenomic Analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef]
- Hsieh, Y.-H.; Peterson, C.M.; Raggio, A.; Keenan, M.J.; Martin, R.J.; Ravussin, E.; Marco, M.L. Impact of Different Fecal Processing Methods on Assessments of Bacterial Diversity in the Human Intestine. Front. Microbiol. 2016, 7, 1643. [Google Scholar] [CrossRef]
- Gorzelak, M.A.; Gill, S.K.; Tasnim, N.; Ahmadi-Vand, Z.; Jay, M.; Gibson, D.L. Methods for Improving Human Gut Microbiome Data by Reducing Variability through Sample Processing and Storage of Stool. PLoS ONE 2015, 10, e0134802. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Marín, C.M.; Isabel Álvarez-Mercado, A.; Plaza-Díaz, J.; Rodríguez-Lara, A.; Gallart-Aragón, T.; Sánchez-Barrón, M.T.; Lartategui, S.D.R.; Alcaide-Lucena, M.; Fernández, M.F.; Fontana, L. A Clustering Study of Sociodemographic Data, Dietary Patterns, and Gut Microbiota in Healthy and Breast Cancer Women Participating in the MICROMA Study. Mol. Nutr. Food Res. 2024, 68, 2400253. [Google Scholar] [CrossRef]
- Hunter, S.J.; Easton, S.; Booth, V.; Henderson, B.; Wade, W.G.; Ward, J.M. Selective Removal of Human DNA from Metagenomic DNA Samples Extracted from Dental Plaque. J. Basic Microbiol. 2011, 51, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in Bacterial Communities along the 2000 Km Salinity Gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A New Perspective for Microbiome Analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Maljkovic Berry, I.; Melendrez, M.C.; Bishop-Lilly, K.A.; Rutvisuttinunt, W.; Pollett, S.; Talundzic, E.; Morton, L.; Jarman, R.G. Next Generation Sequencing and Bioinformatics Methodologies for Infectious Disease Research and Public Health: Approaches, Applications, and Considerations for Development of Laboratory Capacity. J. Infect. Dis. 2019, 221, S292–S307. [Google Scholar] [CrossRef]
- Soni, J.; Sinha, S.; Pandey, R. Understanding Bacterial Pathogenicity: A Closer Look at the Journey of Harmful Microbes. Front. Microbiol. 2024, 15, 1370818. [Google Scholar] [CrossRef]
- Yang, L.; Haidar, G.; Zia, H.; Nettles, R.; Qin, S.; Wang, X.; Shah, F.; Rapport, S.F.; Charalampous, T.; Methé, B.; et al. Metagenomic Identification of Severe Pneumonia Pathogens in Mechanically-Ventilated Patients: A Feasibility and Clinical Validity Study. Respir. Res. 2019, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Moragues-Solanas, L.; Le-Viet, T.; McSorley, E.; Halford, C.; Lockhart, D.S.; Aydin, A.; Kay, G.L.; Elumogo, N.; Mullen, W.; O’Grady, J.; et al. Development and Proof-of-Concept Demonstration of a Clinical Metagenomics Method for the Rapid Detection of Bloodstream Infection. BMC Med. Genom. 2024, 17, 71. [Google Scholar] [CrossRef]
- Mansuri, M.S.; Bathla, S.; Lam, T.T.; Nairn, A.C.; Williams, K.R. Optimal Conditions for Carrying out Trypsin Digestions on Complex Proteomes: From Bulk Samples to Single Cells. J. Proteom. 2024, 297, 105109. [Google Scholar] [CrossRef]
- Li, R.; Liriano, L. A Bone Sample Cleaning Method Using Trypsin for the Isolation of DNA. Leg. Med. 2011, 13, 304–308. [Google Scholar] [CrossRef]
- Han, Y.; Yu, Y.P.; Tseng, G.C.; Luo, J. Trypsin and Reduction Method to Prepare DNA from Formalin-fixed Paraffin-embedded Samples for Methylation Analysis. Histopathology 2009, 54, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.R.; Thompson, L.R. Optimizing an Enclosed Bead Beating Extraction Method for Microbial and Fish Environmental DNA. Environ. DNA 2022, 4, 291–303. [Google Scholar] [CrossRef]
- MetaHIT Consortium (Additional Members); Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Mercado, A.I.; Cano, A.D.V.; Fernández, M.F.; Fontana, L. Gut Microbiota and Breast Cancer: The Dual Role of Microbes. Cancers 2023, 15, 443. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; The Vegan Package. Community Ecology Package. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 3 June 2024).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wen Chen, J.S. RAM-Package: Analysis of Amplicon-Based Metagenomic Data. Available online: https://rdrr.io/cran/RAM/man/RAM-package.html (accessed on 18 June 2020).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||
| Phylum | Healthy Women (Controls) | |||||
| Feces | Breast | |||||
| A (n = 5) | B (n = 5) | C (n = 5) | A (n = 5) | B (n = 5) | C (n = 5) | |
| Actinomycetota | 4.7 (0.3–13.8) a | 7.8 (2.7–10.7) a | 14.4 (13.5–17.5) b | 5.7 (4.8–6.2) | 7.3 (5.6–9.9) | 6.6 (5.0–8.0) |
| Bacteroidota | 6.3 (3.0–22.8) a | 27.2 (15.2–33.9) b | 6.6 (2.6–22.7) a | 16.6 (9.4–19.0) a | 15.9 (6.2–19.2) a | 8.9 (7.1–11.3) b |
| Bacillota | 74.6 (69.7–87.0) a | 62.5 (48.7–72.9) b | 74.1 (63.1–79.6) ab | 57.5 (46.2–63.7) a | 64.6 (52.3–67.6) ab | 71.7 (60.0–78.6) b |
| Fusobacteria | 0 (0–0.03) | 0 (0–0.2) | 0 (0–0) | 0 (0–0.04) | 0 (0–0.1) | 0 (0–0.01) |
| Pseudomonadota | 2.0 (0.2–2.4) a | 0.6 (0.3–1.9) ab | 0.3 (0.1–1.3) b | 4.7 (3.6–10.0) a | 4.8 (3.7–12.6) a | 2.6 (0.5–2.8) b |
| Verrucomicrobiota | 2.3 (0–6.8) a | 0 (0–26.5) b | 0.02 (0–8.0) b | 2.3 (0.8–3.7) | 3.0 (0.2–4.1) | 1.5 (1.1–3.1) |
| Unassigned | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0.04 (0–0.3) | 0 (0–0.5) | 0.09 (0–0.2) |
| (B) | ||||||
| Phylum | Patients with Breast Cancer (Cases) | |||||
| Feces | Breast | |||||
| A (n = 5) | B (n = 5) | A (n = 5) | B (n = 5) | |||
| Actinomycetota | 1.6 (1.1–29.0) | 5.0 (3.5–7.2) | 4.9 (1.9–6.0) | 3.1 (1.5–5.1) | ||
| Bacteroidota | 9.6 (0.8–32.0) a | 17.3 (3.7–24.6) b | 18.3 (16.1–18.9) a | 19.4 (18.4–23.5) b | ||
| Bacillota | 68.2 (52.8–95.0) | 72.4 (58.0–87.7) | 63.0 (57.7–70.2) | 66.9 (60.0–69.2) | ||
| Fusobacteria | 0 (0–0.01) | 0 (0–0) | 0 (0–0.1) | 0.08 (0–0.3) | ||
| Pseudomonadota | 2.0 (0.9–7.7) | 1.2 (0.3–7.6) | 4.6 (3.1–8.2) | 4.2 (2.5–9.1) | ||
| Verrucomicrobiota | 0.9 (0–6.3) a | 5.2 (0.06–14.2) b | 2.3 (1.5–3.0) | 3.0 (1.9–3.5) | ||
| Unassigned | 0 (0–0) | 0 (0–0) | 0.04 (0.02–0.2) | 0.01 (0–0.02) | ||
| Variables | Control | Patients with Breast Cancer | ||||||
|---|---|---|---|---|---|---|---|---|
| Feces | Breast | Feces | Breast | |||||
| A (n = 5) | B (n = 5) | A (n = 5) | B (n = 5) | A (n = 5) | B (n = 5) | A (n = 5) | B (n = 5) | |
| Actinomycetota | 0 (0–21.8) | 1.9 (0–16.0) * | 0 (0–0) | 1.2 (1.0–1.0) | 0.3 (0–26.3) | 6.7 (2.0–14.0) * | 0 (0–0) | 0.7 (1.0–1.0) |
| Bacteroidota | 0 (0–5.5) | 4.2 (0–21.0) * | 0 (0–0) | 10.4 (10.0–10.0) | 5.5 (0–39.2) | 7.7 (1.0–28.0) | 0 (0–0) | 13.4 (12–15) |
| Bacillota | 88.3 (72.6–100) | 83.9 (63–100) | 0 (0–0) | 94.2 (88.0–100.0) | 77.5 (46.8–100) | 83.2 (56–100) | 0 (0–0) | 83.9 (80–88) |
| Pseudomonadota | 0 (0–0) | 1.7 (0–3.0) * | 0 (0–0) | 0 (0–0) | 0 (0–6.7) | 2.7 (1.0–5.0) * | 0 (0–0) | 4.7 (5.0–5.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaza-Díaz, J.; Fernández, M.F.; García, F.; Chueca, N.; Fontana, L.; Álvarez-Mercado, A.I. Comparison of Three DNA Isolation Methods and Two Sequencing Techniques for the Study of the Human Microbiota. Life 2025, 15, 599. https://doi.org/10.3390/life15040599
Plaza-Díaz J, Fernández MF, García F, Chueca N, Fontana L, Álvarez-Mercado AI. Comparison of Three DNA Isolation Methods and Two Sequencing Techniques for the Study of the Human Microbiota. Life. 2025; 15(4):599. https://doi.org/10.3390/life15040599
Chicago/Turabian StylePlaza-Díaz, Julio, Mariana F. Fernández, Federico García, Natalia Chueca, Luis Fontana, and Ana I. Álvarez-Mercado. 2025. "Comparison of Three DNA Isolation Methods and Two Sequencing Techniques for the Study of the Human Microbiota" Life 15, no. 4: 599. https://doi.org/10.3390/life15040599
APA StylePlaza-Díaz, J., Fernández, M. F., García, F., Chueca, N., Fontana, L., & Álvarez-Mercado, A. I. (2025). Comparison of Three DNA Isolation Methods and Two Sequencing Techniques for the Study of the Human Microbiota. Life, 15(4), 599. https://doi.org/10.3390/life15040599