

Genetic and Epigenetic Changes in Arabidopsis thaliana Exposed to Ultraviolet-C Radiation Stress for 25 Generations
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Parental Generation and Progeny Plants
2.2. Plant Growth Conditions
2.3. Lineages Used for UV-Stress Phenotyping
2.4. Phenotypic Analysis Under UV-Stress Treatment
2.5. Primary Root Length Measurements
2.6. Detection of Reactive Oxygen Species
2.7. Lineages Used for Genomic and Epigenomic Studies
2.8. Whole Genome Sequencing (WGS) and Whole Genomic Bisulfite Sequencing (WGBS)
2.9. WGS Library Construction and Sequencing
2.10. WGBS Library Construction
2.11. WGS and WGBS Sequencing
2.12. The Computation and Analysis of Genome Sequence Data
2.13. The Computation and Analysis of WGBS Data
2.14. The Differentially Methylated Regions (DMRs)
2.15. Statistical Analysis and Quality Control Values
3. Results
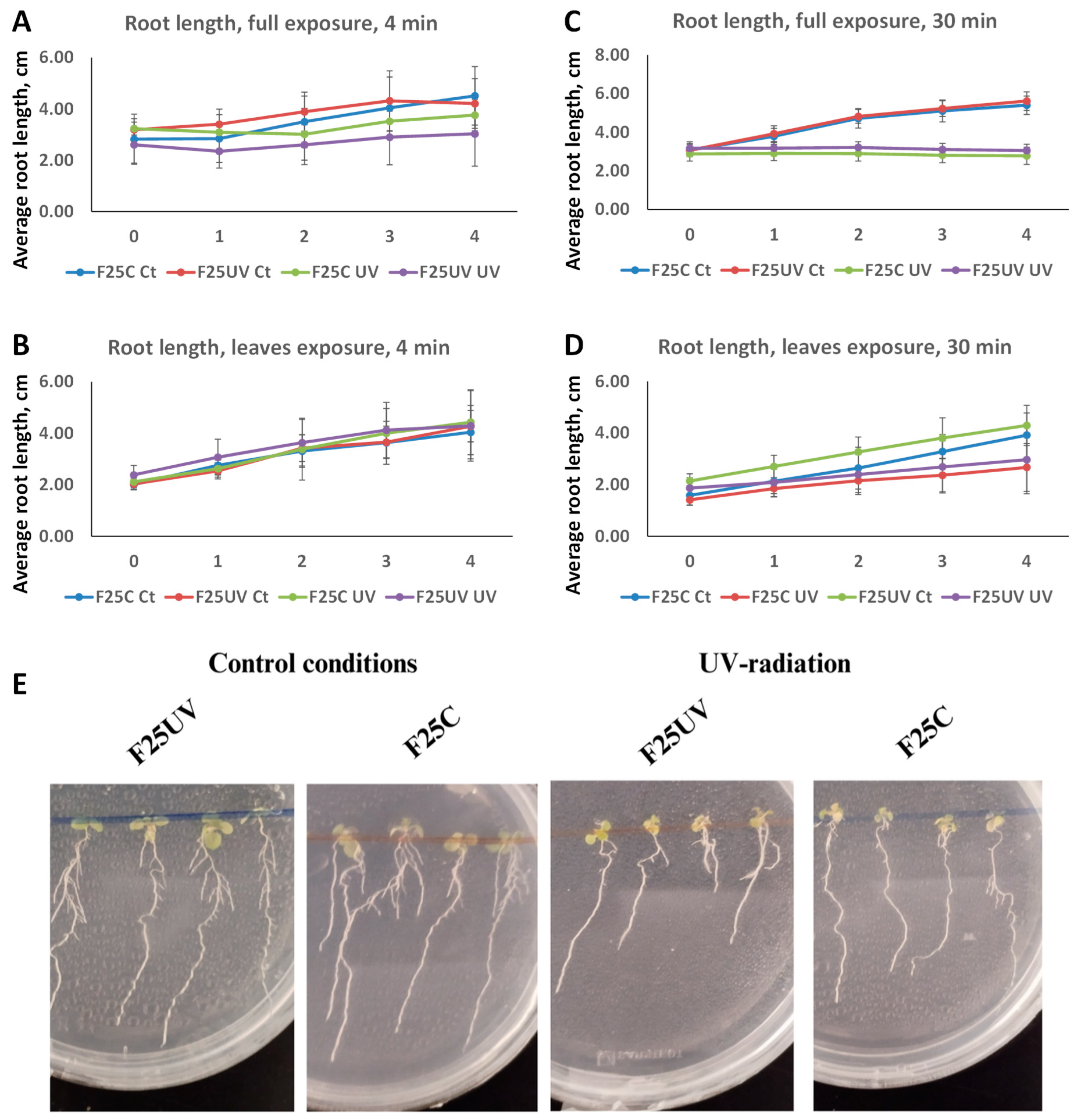
3.1. The Progeny of UV-C-Stressed Plants Did Not Exhibit a UV-Tolerant Phenotype
3.2. Primary Root Elongation Is Inhibited by UV-C Irradiation, Regardless of Transgenerational Inheritance
3.3. F25UV Have Similar Level of Radicals Compared to F25C Plants
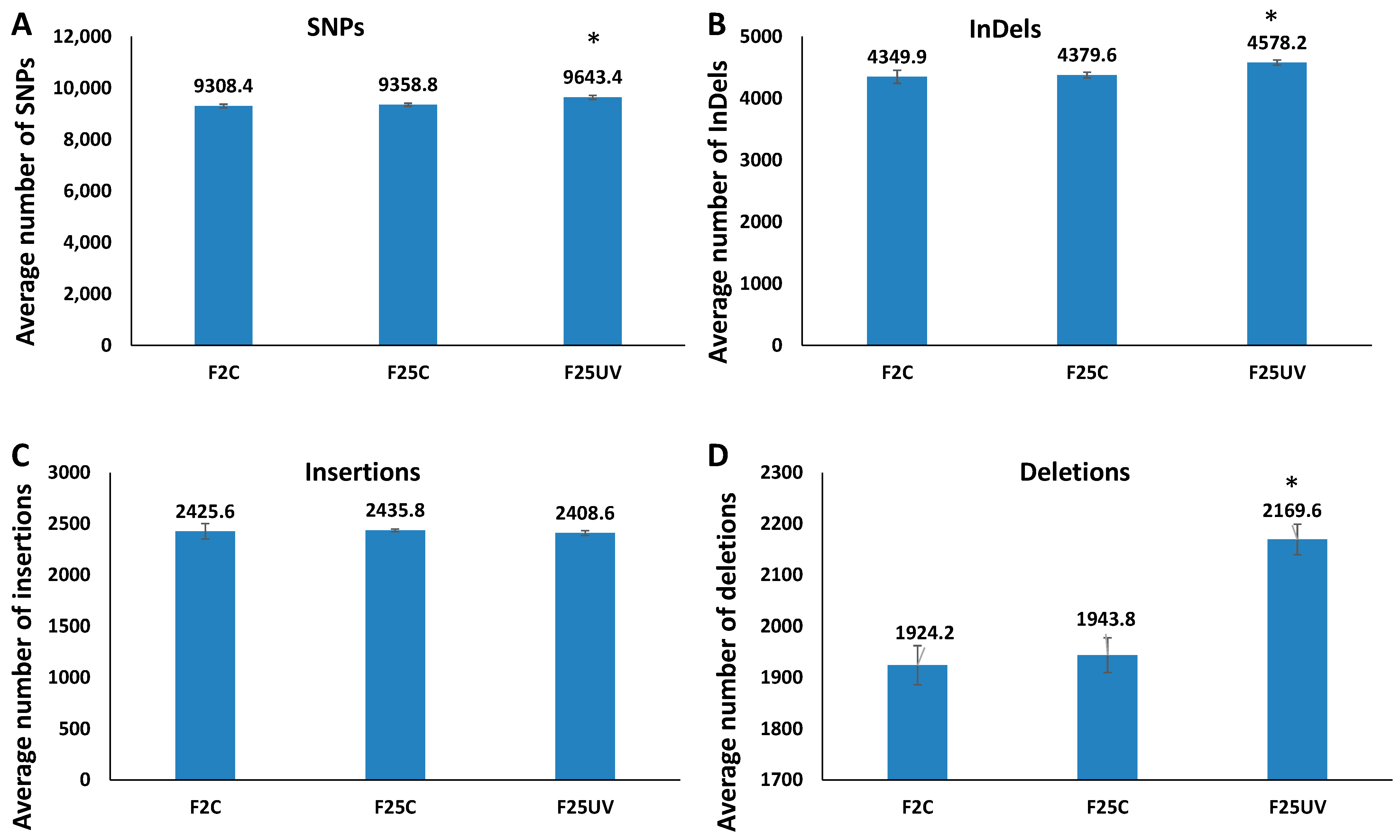
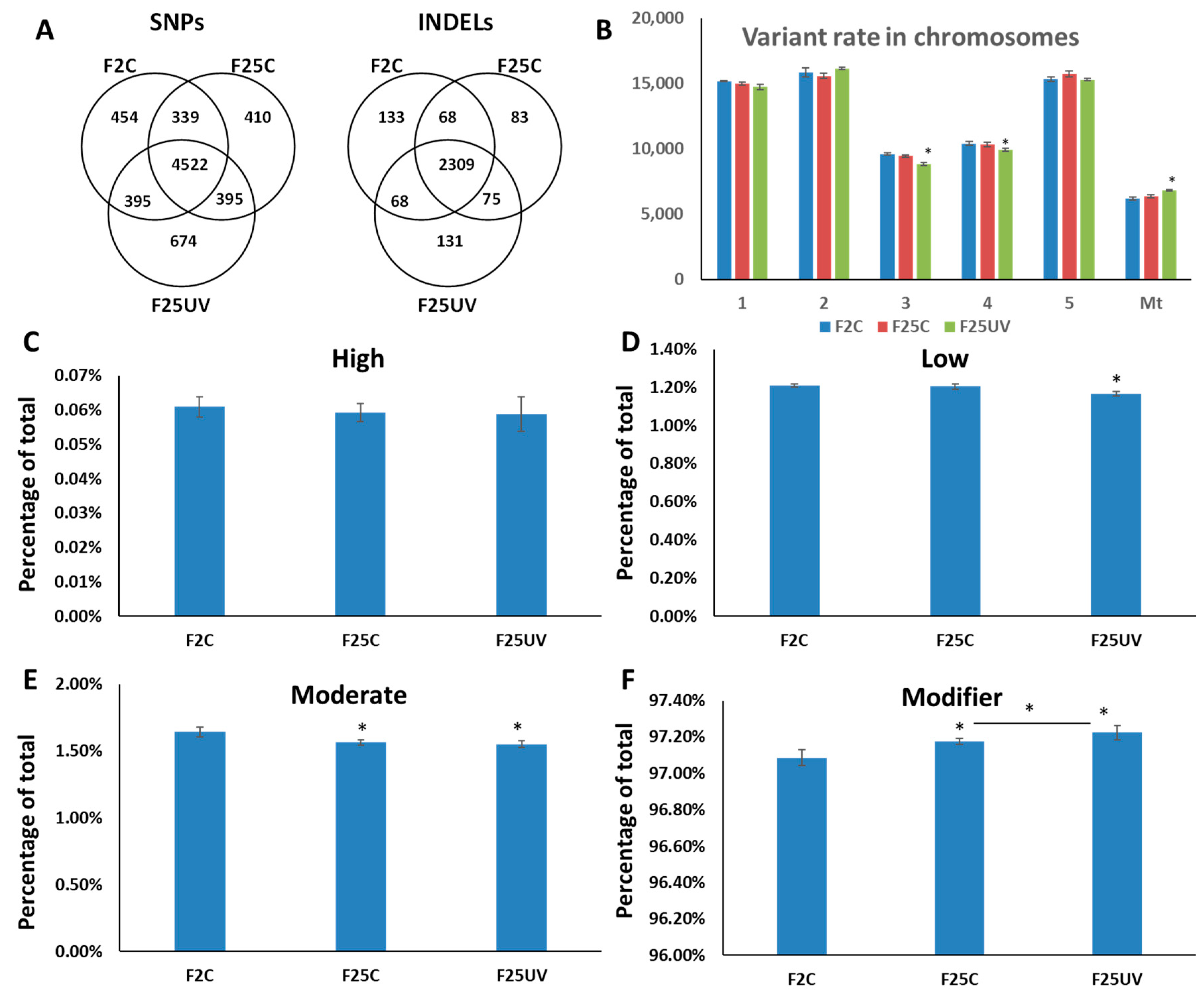
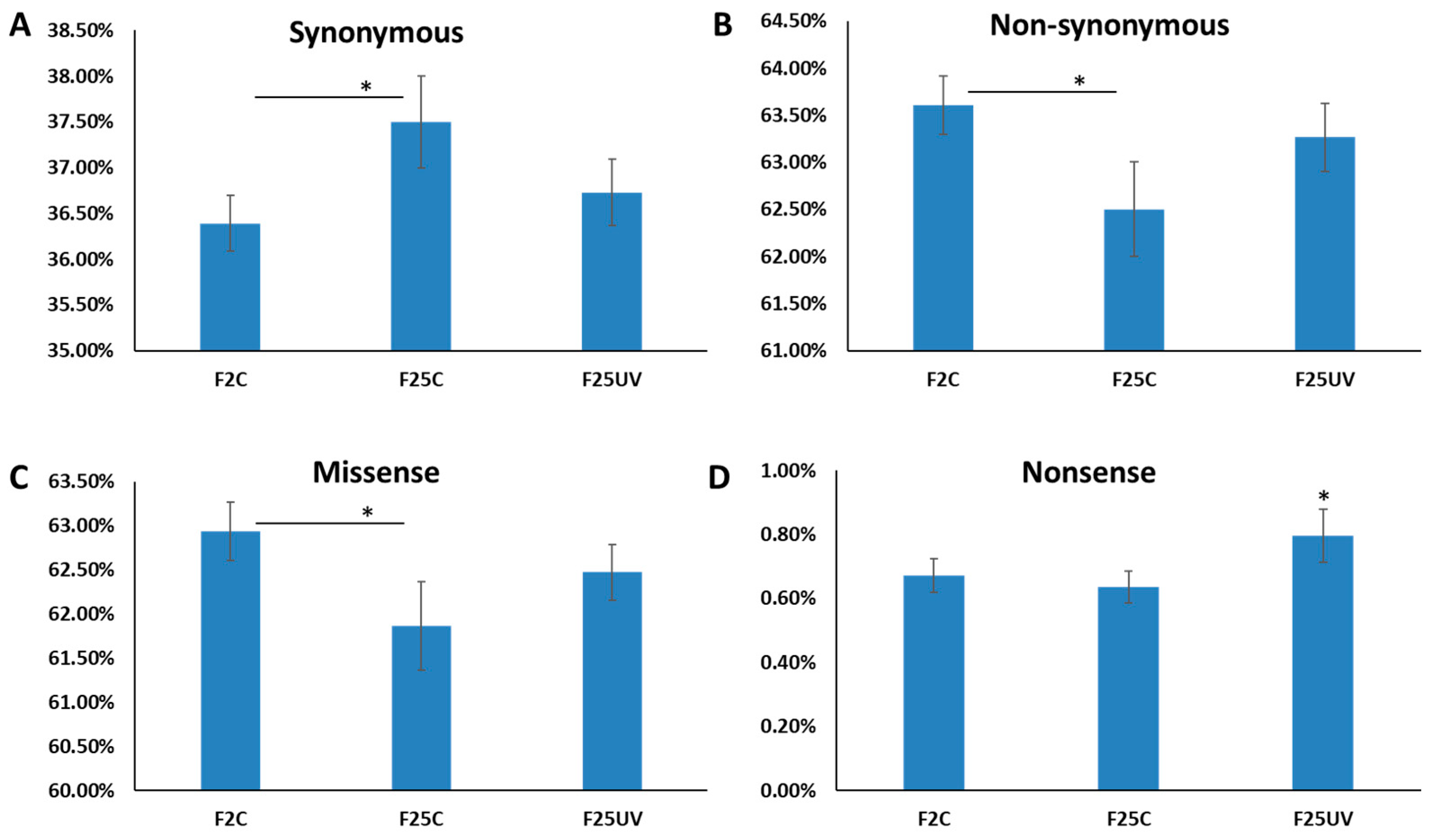
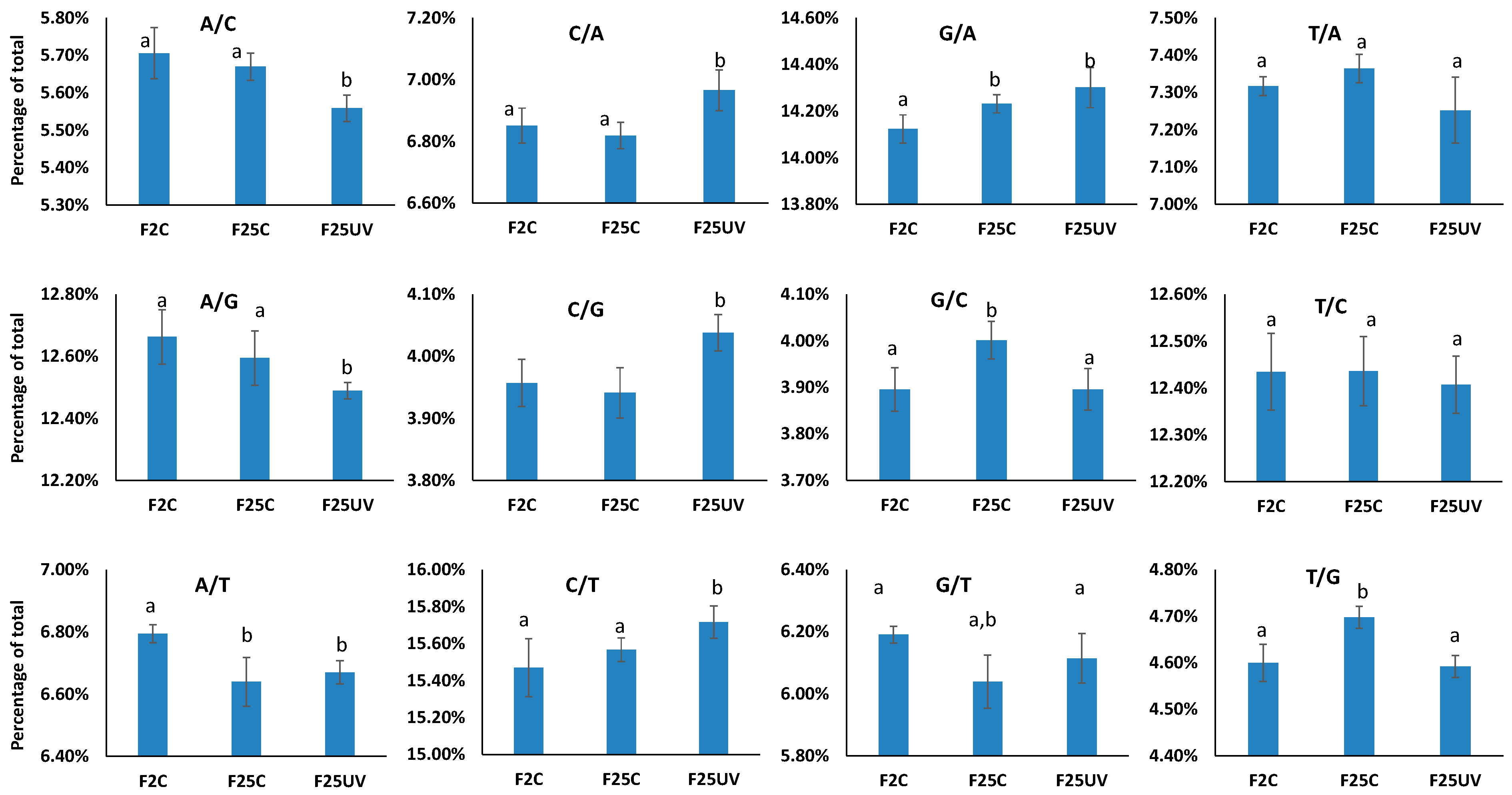
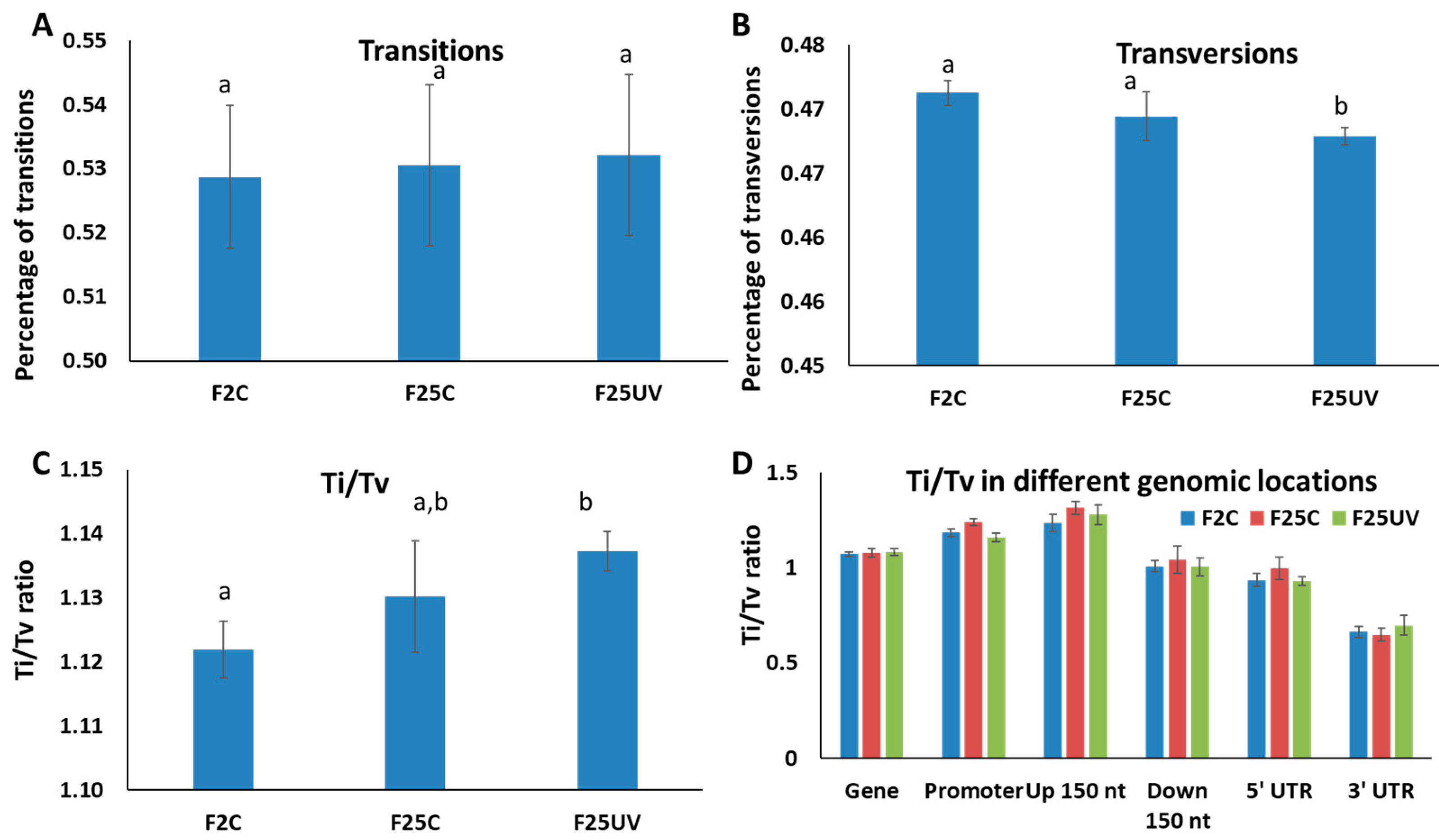
3.4. F25UV Plants Showed a Higher Number of INDELs and Unique SNPs
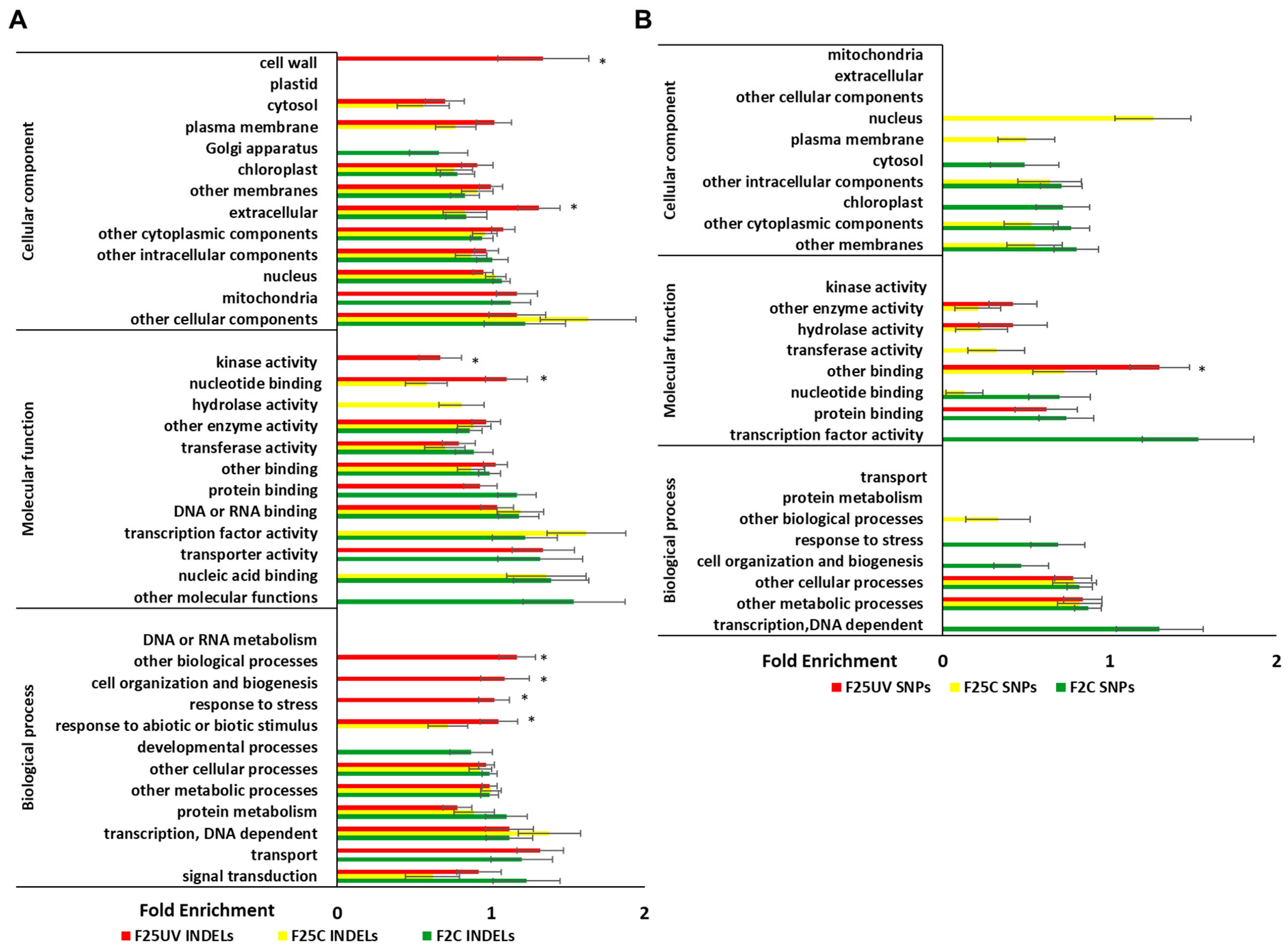
3.5. Genetic Variants in F25UV Were Enriched in Several Cellular Components, Molecular Functions and Biological Processes
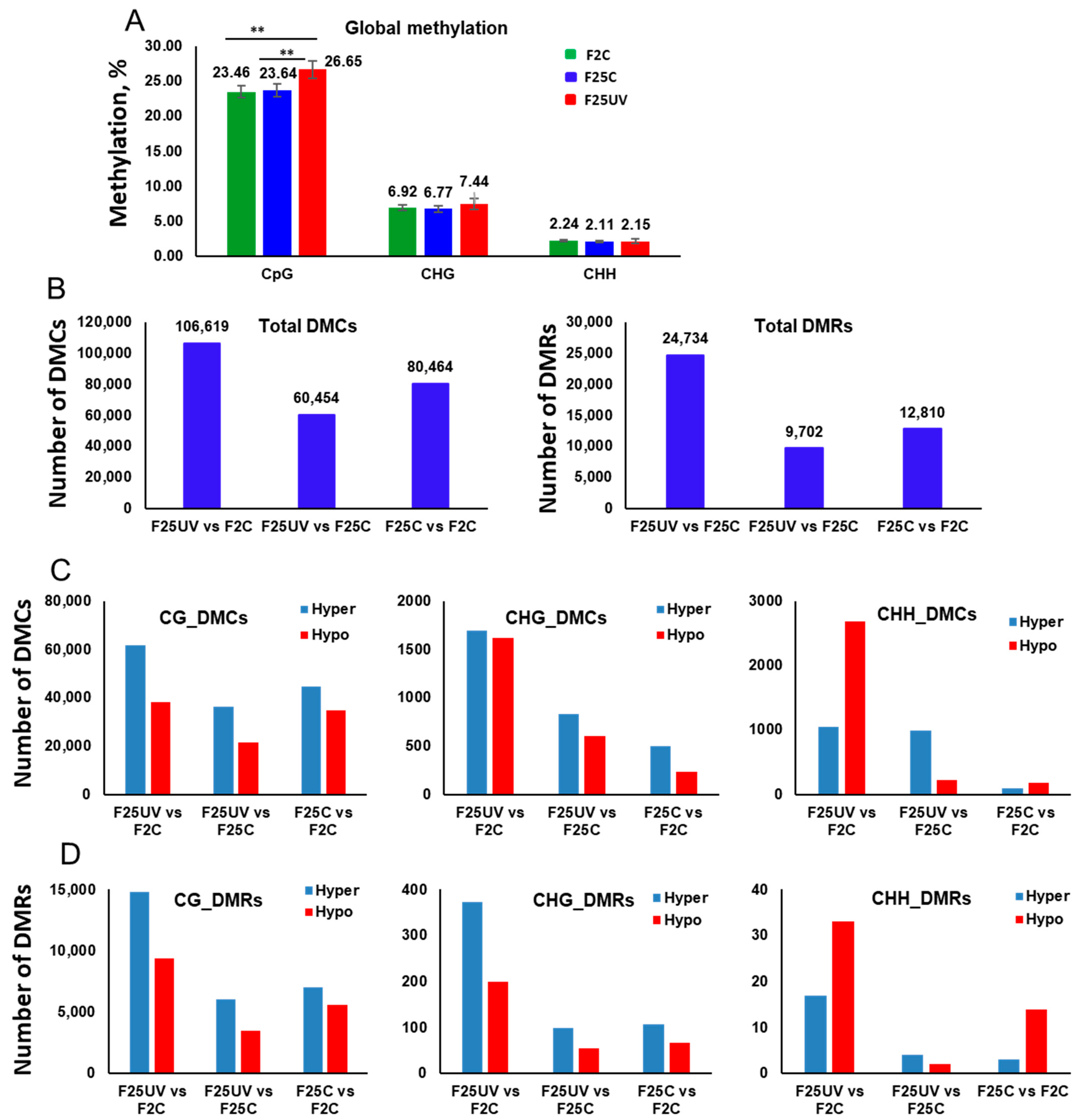
3.6. Multigenerational Exposure to UV-C Increases Global Genome Methylation at the CpG Sites
3.7. The F25UV Group Shows a Higher Number of DMCs and DMRs in Comparison to the F2C Group
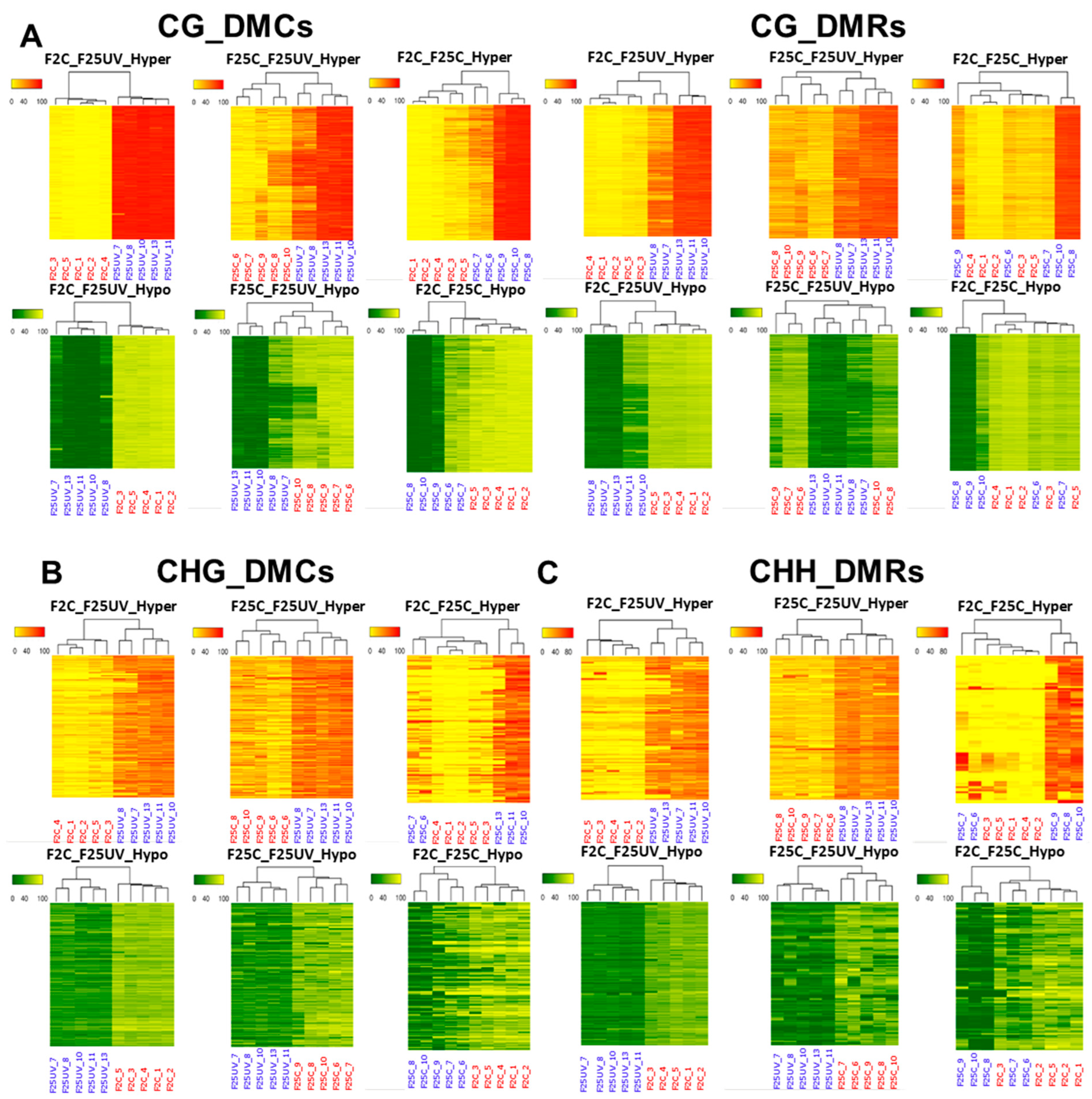
3.8. Plants Belonging to the F25UV and F2C Groups Cluster Separately in All Cytosine Contexts
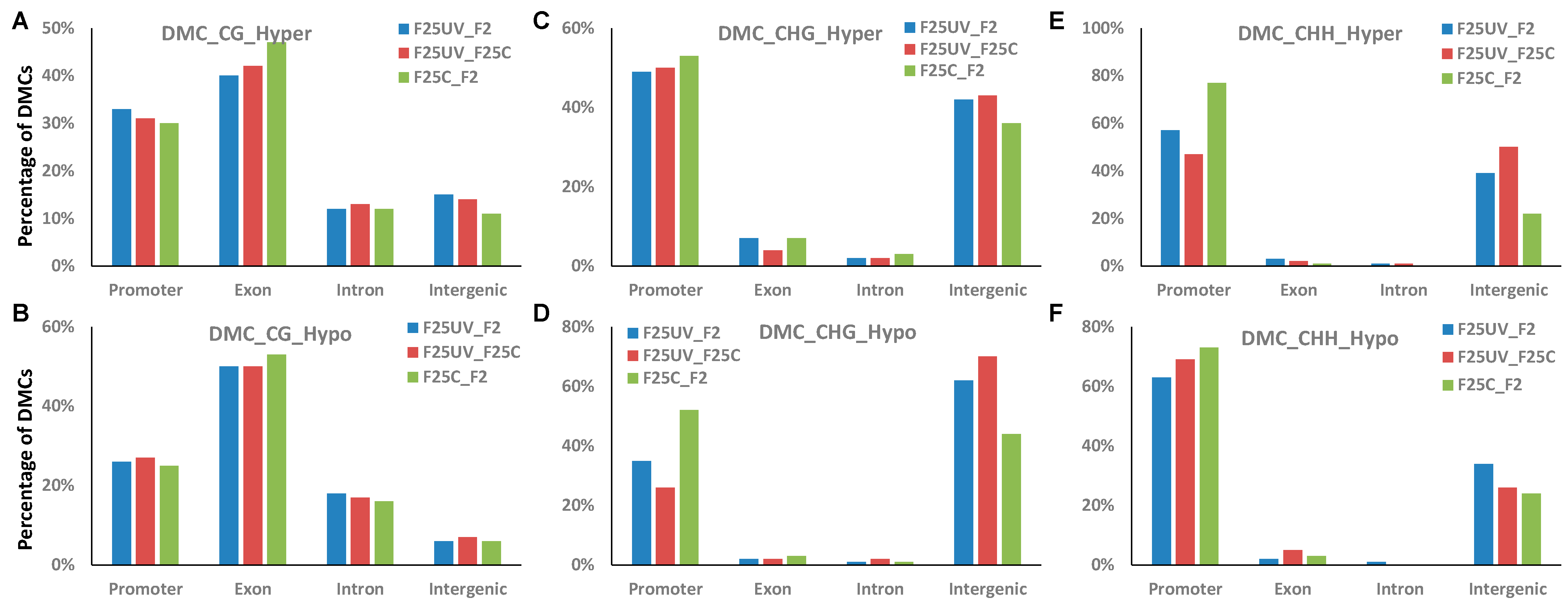
3.9. Analysis of Methylation in the Genic and Intergenic Regions
3.10. Pathway Enrichment for Epimutation-Associated Genes
4. Discussion
4.1. Absence of Phenotypic Resilience to UV-C Stress
4.2. The Analysis of Genetic Variations Induced by Multigenerational Exposure to UV-C
4.3. The Analysis of Epigenetic Variations Induced by Multigenerational Exposure to UV-C
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMC | Differentially methylated cytosine |
| DMR | Differentially methylated region |
| UV-C | Ultraviolet C radiation |
| SNPs | Single nucleotide polymorphisms |
| INDELs | Insertions and deletions |
| CG | Methylation at cytosine followed by guanine |
| CHG | Methylation at cytosine followed by any nucleotide except guanine, followed by guanine |
| CHH | Methylation at any cytosine |
| DCL | Dicer-like protein |
| WGS | Whole genome sequencing |
| WGBS | Whole genome bisulfite sequencing |
| TSS | Transcription start site |
| ROS | Reactive oxygen species |
| F2C | Second generation of progeny of control plants |
| F25C | Twenty-fifth generation of progeny of control plants |
| F25UV | Twenty-fifth generation of UV-stressed plants |
| Tv | Transversions |
| Ti | Transitions |
References
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [PubMed]
- Xavier, M.J.; Roman, S.D.; Aitken, R.J.; Nixon, B. Transgenerational inheritance: How impacts to the epigenetic and genetic information of parents affect offspring health. Hum. Reprod. Update 2019, 25, 518–540. [Google Scholar] [PubMed]
- Abdulraheem, M.I.; Xiong, Y.; Moshood, A.Y.; Cadenas-Pliego, G.; Zhang, H.; Hu, J. Mechanisms of Plant Epigenetic Regulation in Response to Plant Stress: Recent Discoveries and Implications. Plants 2024, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Khan, S.; Wani, I.A.; Gupta, R.; Verma, S.; Alam, P.; Alaklabi, A. Unravelling the Role of Epigenetic Modifications in Development and Reproduction of Angiosperms: A Critical Appraisal. Front. Genet. 2022, 13, 819941. [Google Scholar]
- Quadrana; Colot, V. Plant Transgenerational Epigenetics. Annu. Rev. Genet. 2016, 50, 467–491. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar]
- Iwasaki, M.; Paszkowski, J. Epigenetic memory in plants. EMBO J. 2014, 33, 1987–1998. [Google Scholar]
- Migicovsky, Z.; Yao, Y.; Kovalchuk, I. Transgenerational phenotypic and epigenetic changes in response to heat stress in Arabidopsis thaliana. Plant Signal Behav. 2014, 9, e27971. [Google Scholar]
- Yadav, N.S.; Titov, V.; Ayemere, I.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Multigenerational Exposure to Heat Stress Induces Phenotypic Resilience, and Genetic and Epigenetic Variations in Arabidopsis thaliana Offspring. Front. Plant Sci. 2022, 13, 728167. [Google Scholar]
- Rahman, A.; Yadav, N.S.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants. Int. J. Mol. Sci. 2024, 25, 2795. [Google Scholar] [CrossRef]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar]
- Bilichak, A.; Ilnystkyy, Y.; Hollunder, J.; Kovalchuk, I. The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 2012, 7, e30515. [Google Scholar]
- Migicovsky, Z.; Kovalchuk, I. Transgenerational changes in plant physiology and in transposon expression in response to UV-C stress in Arabidopsis thaliana. Plant Signal Behav. 2014, 9, e976490. [Google Scholar] [PubMed]
- Daudi, A.; O’Brien, J.A. Detection of Hydrogen Peroxide by DAB Staining in Arabidopsis Leaves. Bio Protoc. 2012, 2, e263. [Google Scholar]
- Bustos, D.; Lascano, R.; Villasuso, A.L.; Machado, E.; Senn, M.E.; Córdoba, A.; Taleisnik, E. Reductions in maize root-tip elongation by salt and osmotic stress do not correlate with apoplastic O2*-levels. Ann. Bot. 2008, 102, 551–559. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar]
- Akalin, A.; Kormaksson, M.; Li, S.; E Garrett-Bakelman, F.; E Figueroa, M.; Melnick, A.; E Mason, C. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar]
- Provart, N.J.; Zhu, T. A browser-based functional classification SuperViewer for Arabidopsis genomics. Curr. Comp. Mol. Biol. 2003, 2003, 271–272. [Google Scholar]
- Sheridan, M.L.; Simonelli, L.; Giustozzi, M.; Casati, P. Ultraviolet-B Radiation Represses Primary Root Elongation by Inhibiting Cell Proliferation in the Meristematic Zone of Arabidopsis Seedlings. Front. Plant Sci. 2022, 13, 829336. [Google Scholar]
- Zhang, L.; Wang, X.; Zu, Y.; He, Y.; Li, Z.; Li, Y. Effects of UV-B Radiation Exposure on Transgenerational Plasticity in Grain Morphology and Proanthocyanidin Content in Yuanyang Red Rice. Int. J. Mol. Sci. 2024, 25, 4766. [Google Scholar] [CrossRef]
- Rahavi, M.R.; Migicovsky, Z.D.; Titov, V.; Kovalchuk, I. Transgenerational adaptation to heavy metal salts in Arabidopsis. Front. Plant Sci. 2011, 2, 91. [Google Scholar]
- Pecinka, A.; Rosa, M.; Schikora, A.; Berlinger, M.; Hirt, H.; Luschnig, C.; Scheid, O.M. Transgenerational stress memory is not a general response in Arabidopsis. PLoS ONE 2009, 4, e5202. [Google Scholar]
- Shahmohamadloo, R.S.; Fryxell, J.M.; Rudman, S.M. Transgenerational epigenetic inheritance increases trait variation but is not adaptive. Evolution 2025, qpaf050. [Google Scholar] [CrossRef]
- Yun, S.H.; Noh, B.; Noh, Y.S. Negative evidence on the transgenerational inheritance of defense priming in Arabidopsis thaliana. BMB Rep. 2022, 55, 342–347. [Google Scholar]
- Van Dooren, T.J.M.; Silveira, A.B.; Gilbault, E.; Jiménez-Gómez, J.M.; Martin, A.; Bach, L.; Tisné, S.; Quadrana, L.; Loudet, O.; Colot, V. Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 2020, 71, 3588–3602. [Google Scholar]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and Its consequences. In Experientia Supplementum; Birkhäuser: Basel, Switzerland, 2009; Volume 99, pp. 87–110. [Google Scholar]
- Kim, J.-H.; Ryu, T.H.; Lee, S.S.; Lee, S.; Chung, B.Y. Ionizing radiation manifesting DNA damage response in plants: An overview of DNA damage signaling and repair mechanisms in plants. Plant Sci. 2019, 278, 44–53. [Google Scholar]
- Dutta, S.; Mitra, M.; Agarwal, P.; Mahapatra, K.; De, S.; Sett, U.; Roy, S. Oxidative and genotoxic damages in plants in response to heavy metal stress and maintenance of genome stability. Plant Signal Behav. 2018, 13, e1460048. [Google Scholar]
- Arrieta, M.; Willems, G.; DePessemier, J.; Colas, I.; Burkholz, A.; Darracq, A.; Vanstraelen, S.; Pacolet, P.; Barré, C.; Kempeneers, P.; et al. The effect of heat stress on sugar beet recombination. Theor. Appl. Genet. 2021, 134, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Kovalchuk, I. Abiotic stress leads to somatic and heritable changes in homologous recombination frequency, point mutation frequency and microsatellite stability in Arabidopsis plants. Mutat. Res. 2011, 707, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.; Singh, A.K.; Dhanapal, S.; Saeed, T.H.; Hyde, G.J.; Baskar, R. Brief temperature stress during reproductive stages alters meiotic recombination and somatic mutation rates in the progeny of Arabidopsis. BMC Plant Biol. 2017, 17, 103. [Google Scholar] [CrossRef]
- Lu, Z.; Cui, J.; Wang, L.; Teng, N.; Zhang, S.; Lam, H.-M.; Zhu, Y.; Xiao, S.; Ke, W.; Lin, J.; et al. Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures. Genome Biol. 2021, 22, 160. [Google Scholar] [CrossRef]
- Yao, Y.; Bilichak, A.; Titov, V.; Golubov, A.; Kovalchuk, I. Genome stability of Arabidopsis atm, ku80 and rad51b mutants: Somatic and transgenerational responses to stress. Plant Cell Physiol. 2013, 54, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration memory of stress in plants. Nature 2006, 442, 1046–1049. [Google Scholar] [CrossRef]
- Nakamura, M.; Nunoshiba, T.; Hiratsu, K. Detection and analysis of UV-induced mutations in the chromosomal DNA of Arabidopsis. Biochem. Biophys. Res. Commun. 2021, 554, 89–93. [Google Scholar] [CrossRef]
- Xia, J.; Han, L.; Zhao, Z. Investigating the relationship of DNA methylation with mutation rate and allele frequency in the human genome. BMC Genom. 2012, 13 (Suppl. S8), S7. [Google Scholar] [CrossRef]
- Cooper, D.N.; Mort, M.; Stenson, P.D.; Ball, E.V.; A Chuzhanova, N. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum. Genom. 2010, 4, 406–410. [Google Scholar] [CrossRef]
- Stoltzfus, A.; McCandlish, D.M. Mutational Biases Influence Parallel Adaptation. Mol. Biol. Evol. 2017, 34, 2163–2172. [Google Scholar] [CrossRef]
- Storz, J.F.; Natarajan, C.; Signore, A.V.; Witt, C.C.; McCandlish, D.M.; Stoltzfus, A. The role of mutation bias in adaptive molecular evolution: Insights from convergent changes in protein function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180238. [Google Scholar]
- Pértille, F.; Da Silva, V.H.; Johansson, A.M.; Lindström, T.; Wright, D.; Coutinho, L.L.; Jensen, P.; Guerrero-Bosagna, C. Mutation dynamics of CpG dinucleotides during a recent event of vertebrate diversification. Epigenetics 2019, 14, 685–707. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. Elife 2016, 5, e13546. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.-T.; Aufsatz, W.; Jonak, C.; Luschnig, C. Transgenerational epigenetic inheritance in plants. Biochim. Biophys. Acta 2011, 1809, 459–468. [Google Scholar] [PubMed]
- Luna, E.; Bruce, T.J.; Roberts, M.R.; Flors, V.; Ton, J. Next-generation systemic acquired resistance. Plant Physiol. 2012, 158, 844–853. [Google Scholar] [CrossRef]
- Ohlsson, A.B.; Segerfeldt, P.; Lindström, A.; Borg-Karlson, A.-K.; Berglund, T. UV-B exposure of indoor-grown Picea abies seedlings causes an epigenetic effect and selective emission of terpenes. Z. Naturforsch C J. Biosci. 2013, 68, 139–147. [Google Scholar]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Cortijo, S.; Wardenaar, R.; Colomé-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.-M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef]
- Laanen, P.; Saenen, E.; Mysara, M.; Van de Walle, J.; Van Hees, M.; Nauts, R.; Van Nieuwerburgh, F.; Voorspoels, S.; Jacobs, G.; Cuypers, A.; et al. Changes in DNA Methylation in Arabidopsis thaliana Plants Exposed Over Multiple Generations to Gamma Radiation. Front. Plant Sci. 2021, 12, 611783. [Google Scholar]
- Youngson, N.A.; Lin, P.C.; Lin, S.S. The convergence of autophagy, small RNA and the stress response—Implications for transgenerational epigenetic inheritance in plants. Biomol. Concepts 2014, 5, 1–8. [Google Scholar] [PubMed]
- Bilichak, A.; Ilnytskyy, Y.; Wóycicki, R.; Kepeshchuk, N.; Fogen, D.; Kovalchuk, I.; Wóycicki, R. The elucidation of stress memory inheritance in Brassica rapa plants. Front. Plant Sci. 2015, 6, 5. [Google Scholar]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar]
- Takuno, S.; Gaut, B.S. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol. Biol. Evol. 2012, 29, 219–227. [Google Scholar]
- Horvath, R.; Laenen, B.; Takuno, S.; Slotte, T. Single-cell expression noise and gene-body methylation in Arabidopsis thaliana. Heredity 2019, 123, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Muyle, A.M.; Seymour, D.K.; Lv, Y.; Huettel, B.; Gaut, B.S. Gene Body Methylation in Plants: Mechanisms, Functions, and Important Implications for Understanding Evolutionary Processes. Genome Biol. Evol. 2022, 14, evac038. [Google Scholar]
- van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [PubMed]
- Briffa, A.; Hollwey, E.; Shahzad, Z.; Moore, J.D.; Lyons, D.B.; Howard, M.; Zilberman, D. Millennia-long epigenetic fluctuations generate intragenic DNA methylation variance in Arabidopsis populations. Cell Syst. 2023, 14, 953–967.e17. [Google Scholar]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar]
- Jiang, C.; Mithani, A.; Belfield, E.J.; Mott, R.; Hurst, L.D.; Harberd, N.P. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 2014, 24, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt Tolerant and Sensitive Rice Varieties Display Differential Methylome Flexibility under Salt Stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef] [PubMed]
- Dhanya Thomas, T.T.; Dinakar, C.; Puthur, J.T. Effect of UV-B priming on the abiotic stress tolerance of stress-sensitive rice seedlings: Priming imprints and cross-tolerance. Plant Physiol. Biochem. 2020, 147, 21–30. [Google Scholar] [CrossRef]
- Alamer, K.H.; Attia, H. UV-C seed priming improves tomato plant growth against salt stress. J. Taibah Uni. Sci. 2022, 16, 1181–1191. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virgen, A.L.; Yadav, N.S.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Genetic and Epigenetic Changes in Arabidopsis thaliana Exposed to Ultraviolet-C Radiation Stress for 25 Generations. Life 2025, 15, 502. https://doi.org/10.3390/life15030502
Virgen AL, Yadav NS, Byeon B, Ilnytskyy Y, Kovalchuk I. Genetic and Epigenetic Changes in Arabidopsis thaliana Exposed to Ultraviolet-C Radiation Stress for 25 Generations. Life. 2025; 15(3):502. https://doi.org/10.3390/life15030502
Chicago/Turabian StyleVirgen, Andres Lopez, Narendra Singh Yadav, Boseon Byeon, Yaroslav Ilnytskyy, and Igor Kovalchuk. 2025. "Genetic and Epigenetic Changes in Arabidopsis thaliana Exposed to Ultraviolet-C Radiation Stress for 25 Generations" Life 15, no. 3: 502. https://doi.org/10.3390/life15030502
APA StyleVirgen, A. L., Yadav, N. S., Byeon, B., Ilnytskyy, Y., & Kovalchuk, I. (2025). Genetic and Epigenetic Changes in Arabidopsis thaliana Exposed to Ultraviolet-C Radiation Stress for 25 Generations. Life, 15(3), 502. https://doi.org/10.3390/life15030502