
Developmental Proteomics Reveals the Dynamic Expression Profile of Global Proteins of Haemaphysalis longicornis (Parthenogenesis)

Abstract

1. Introduction
2. Materials and Methods
2.1. Rabbits and Parasites
2.2. The Collection of H. longicornis at Different Developmental Stages
2.3. Protein Extraction
2.4. Proteolysis and Peptide Labeling
2.5. LC-MS/MS Analysis
2.6. Database Search and Bioinformatics Analysis
2.7. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR) Analysis
2.8. Statistical Analysis
3. Results and Discussion
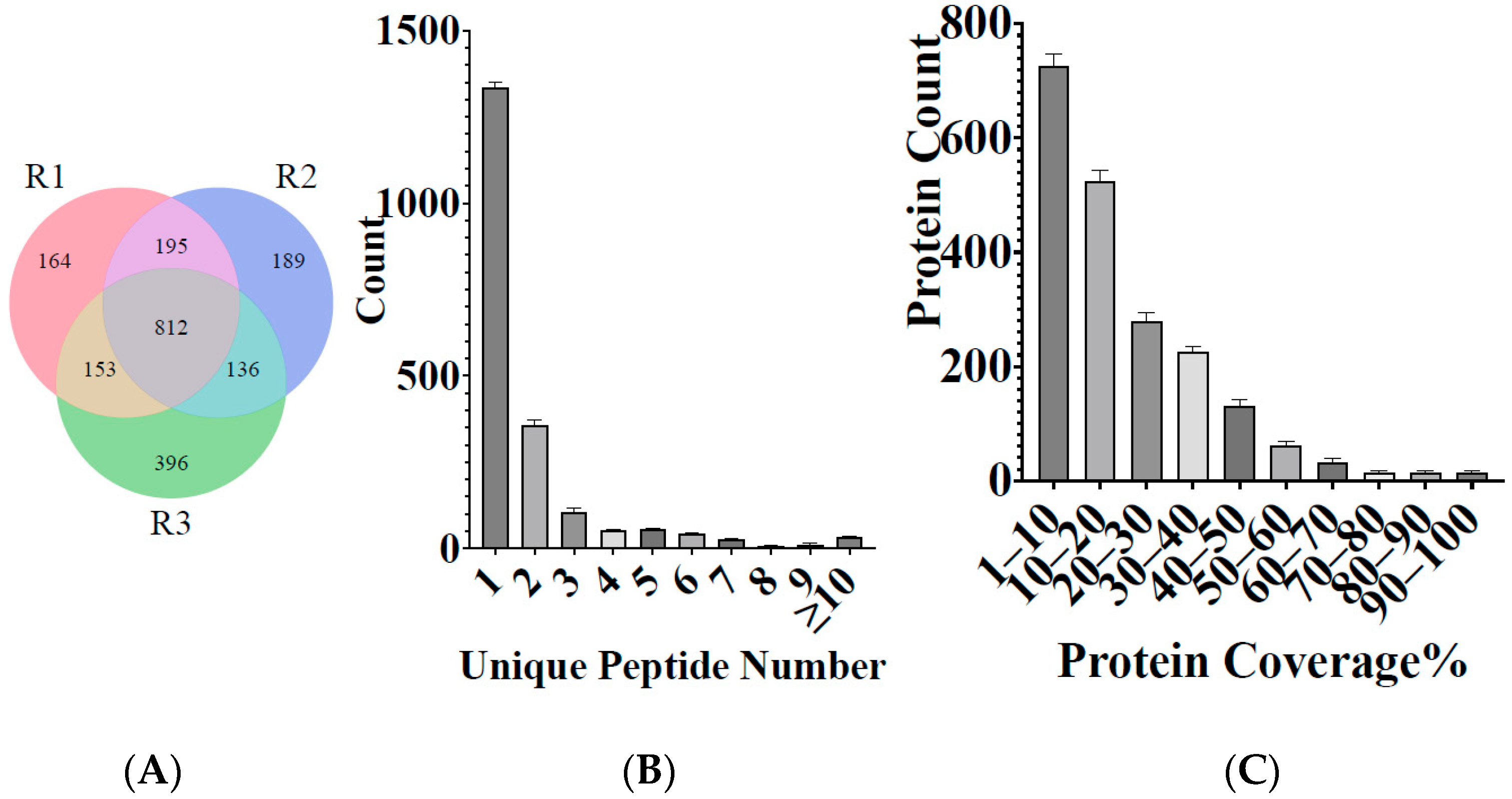
3.1. Overview of Main Data and Protein Identification
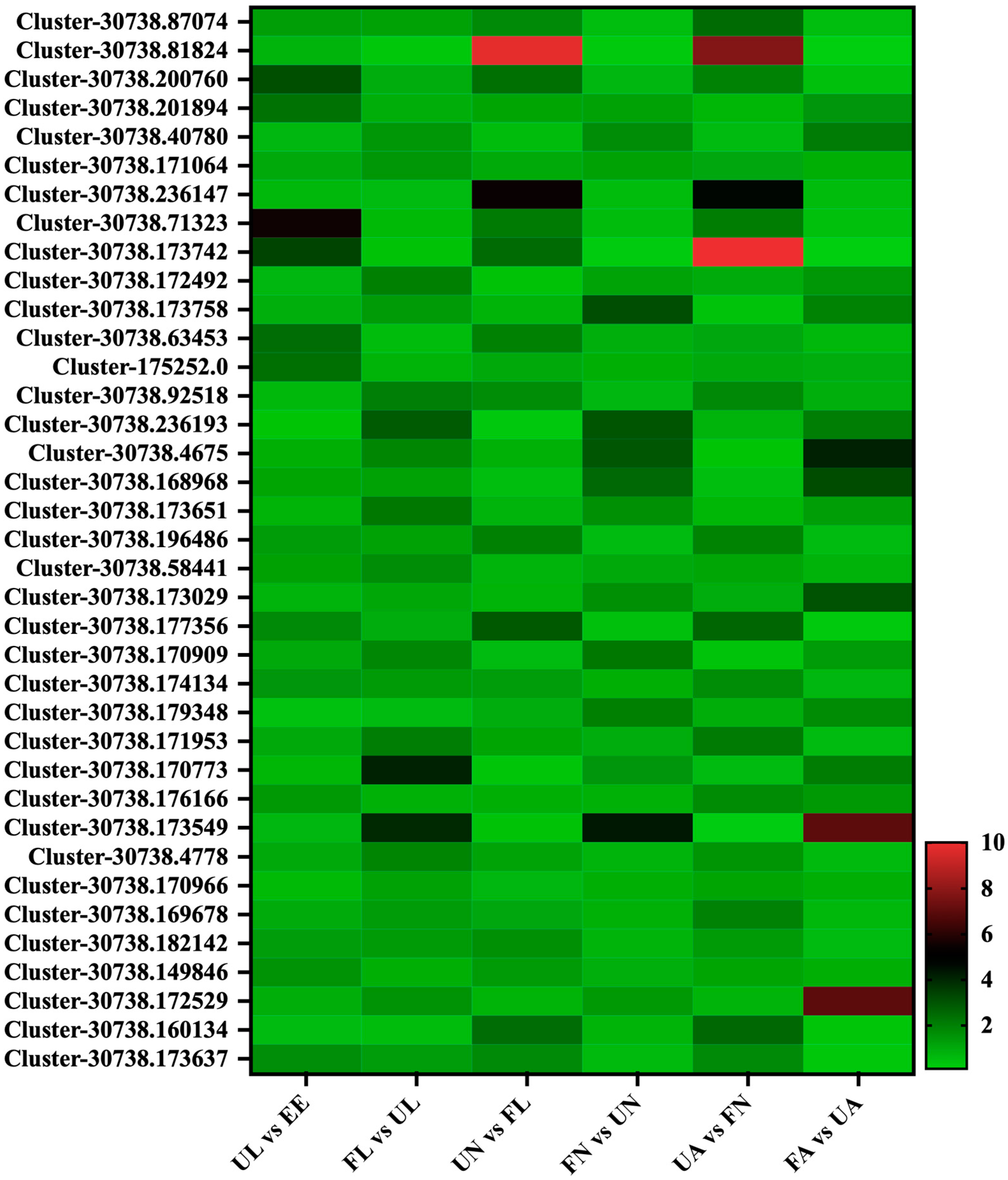
3.2. Protein Quantification
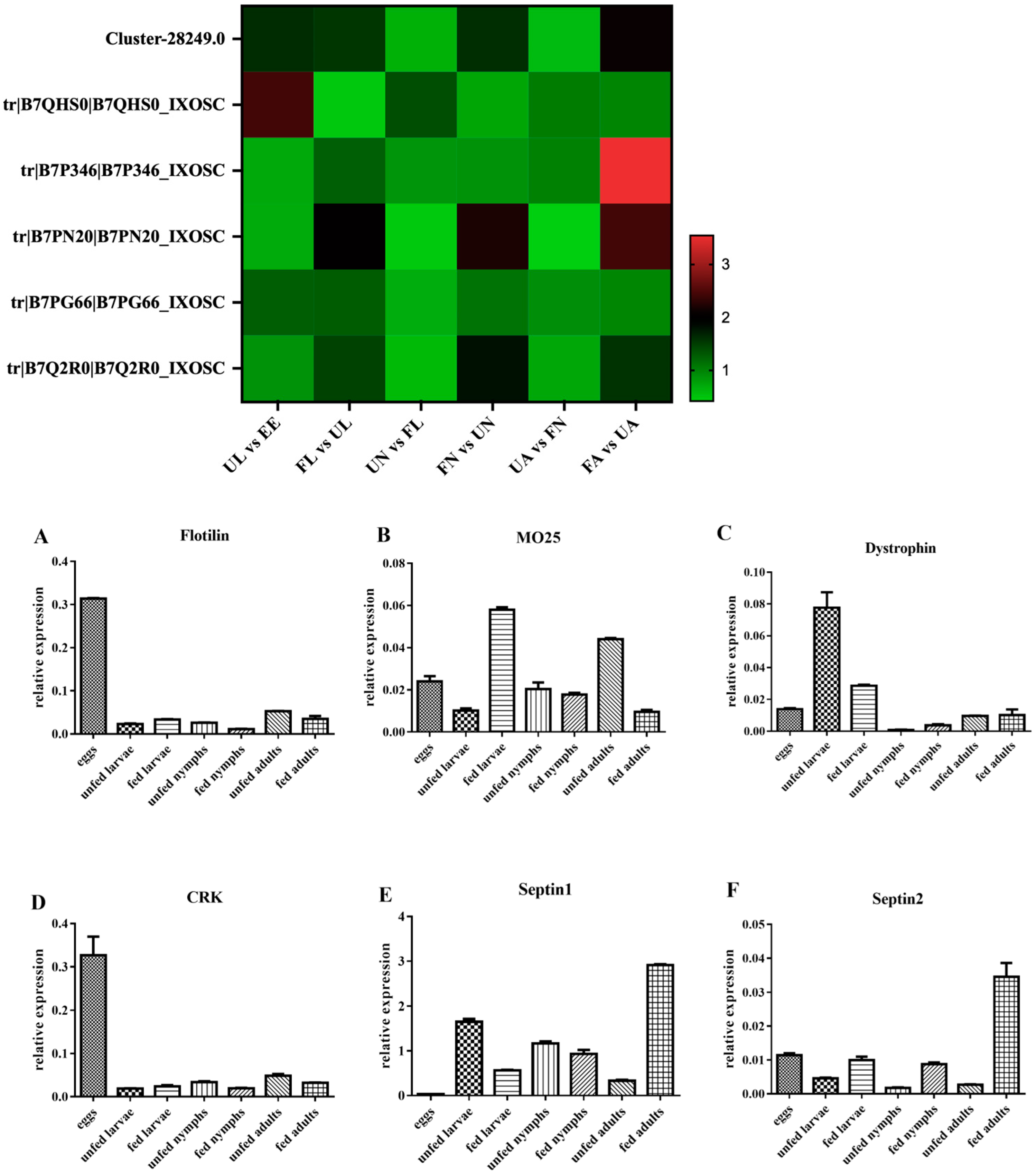
3.3. RT-qPCR Analysis
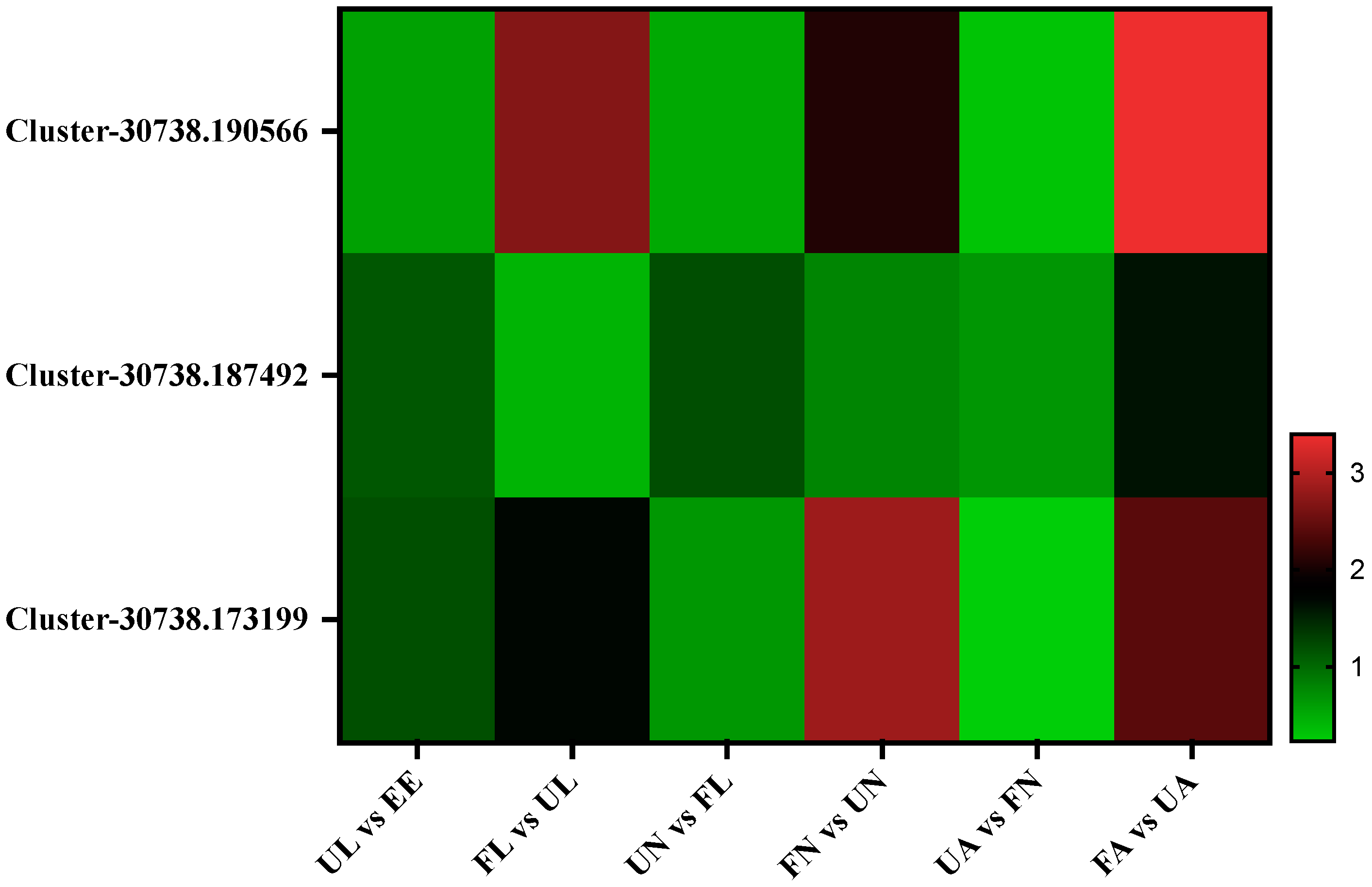
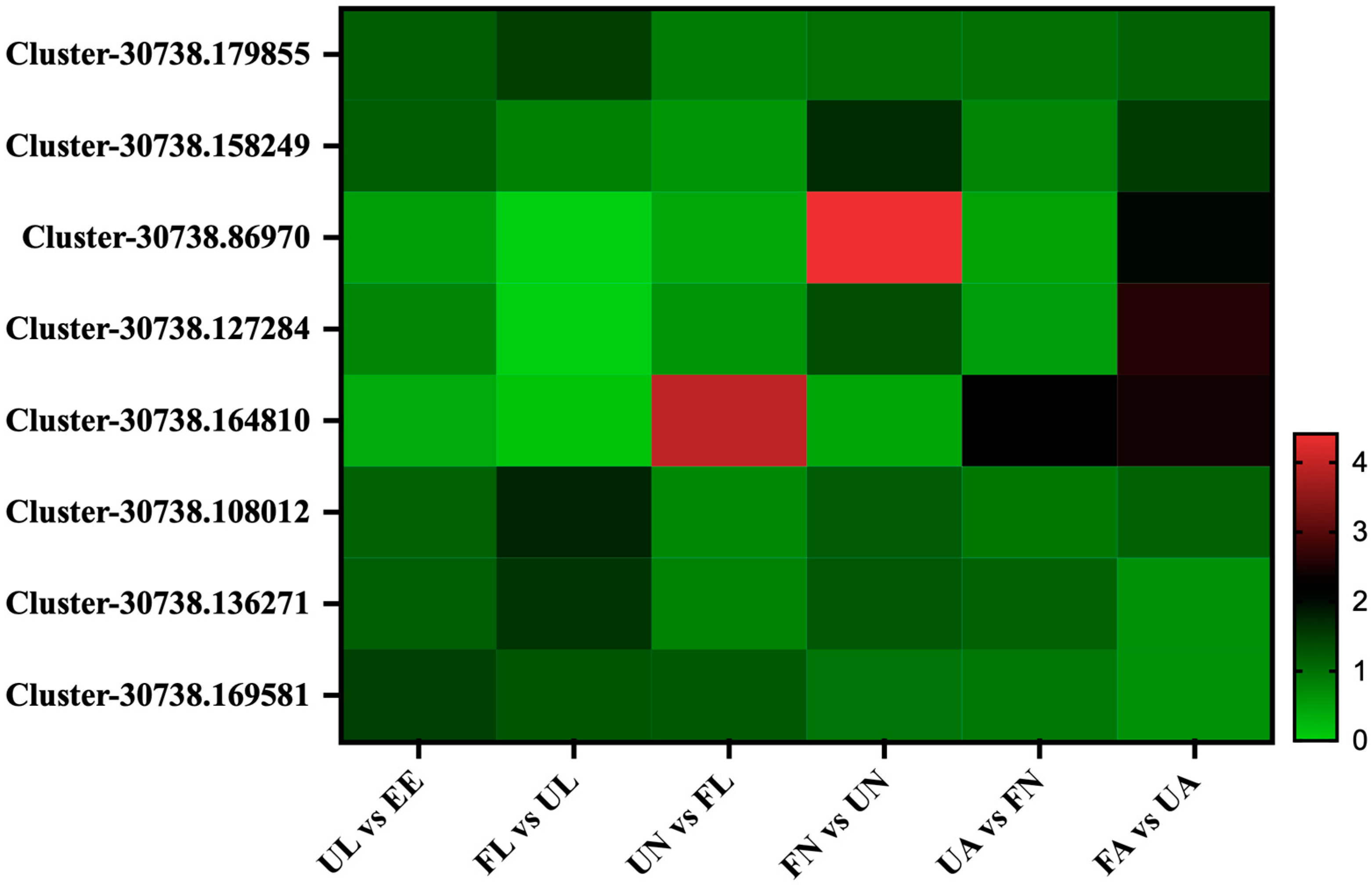
3.4. Expression Profile of the Identified Proteins
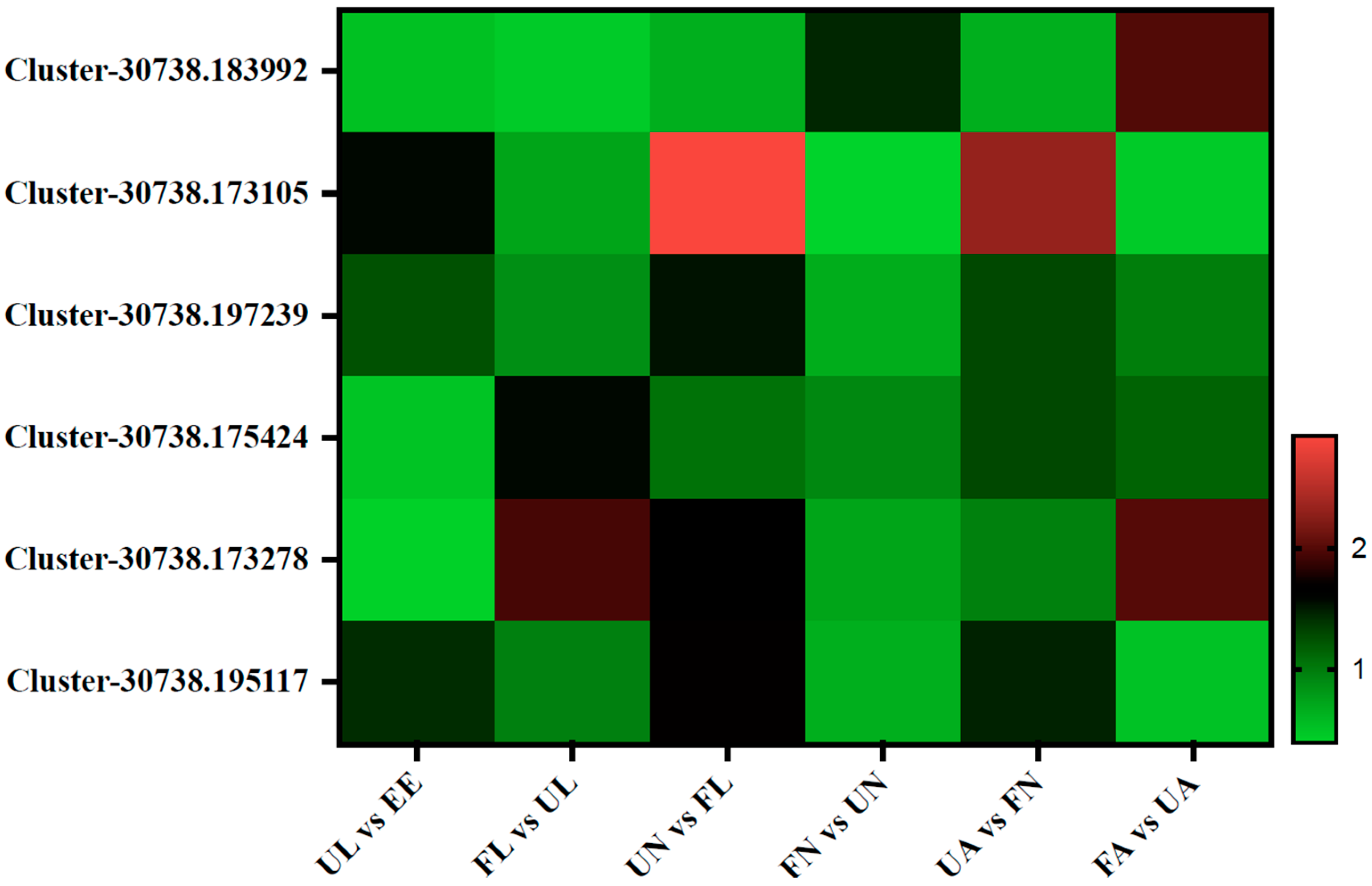
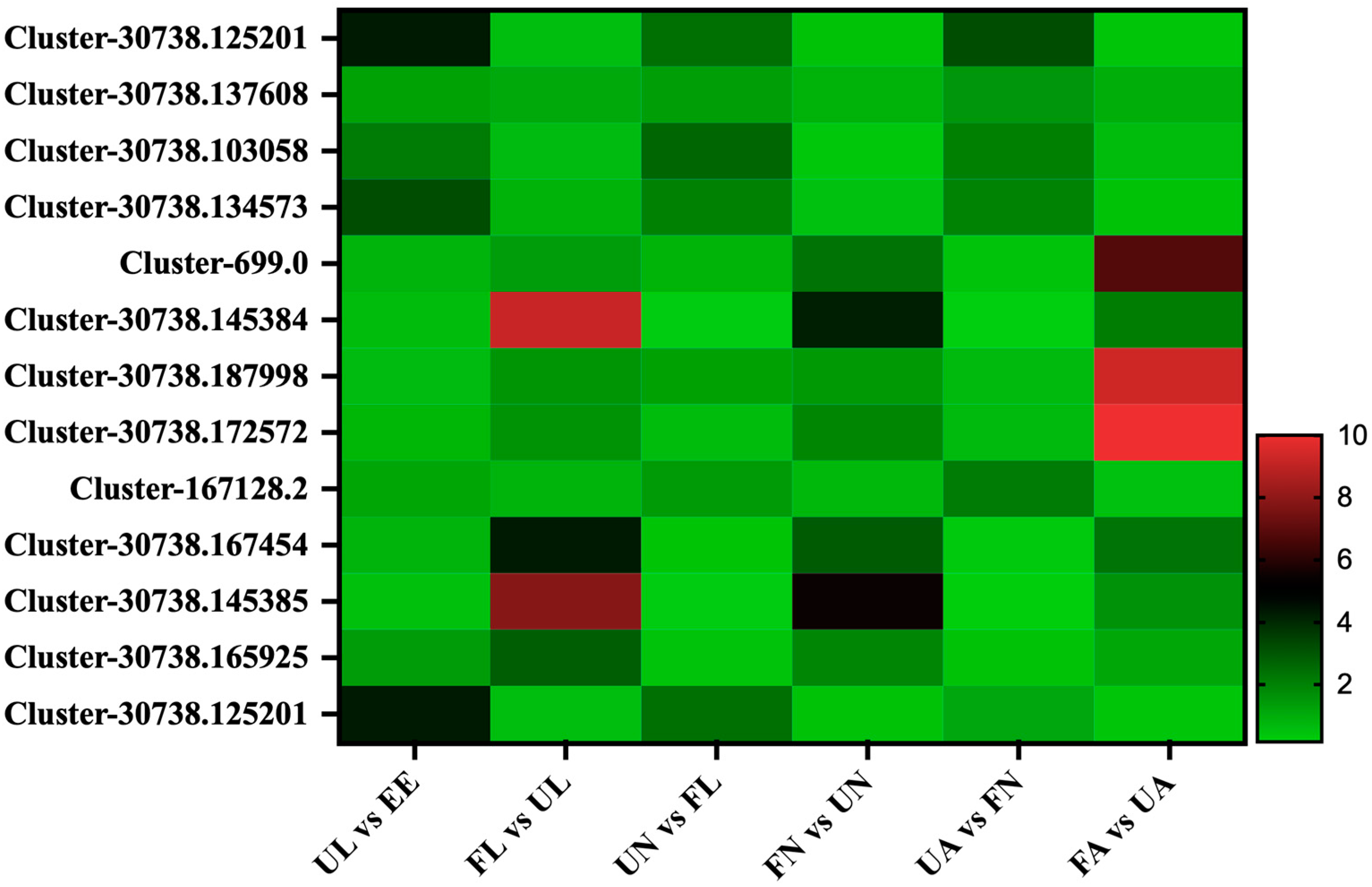
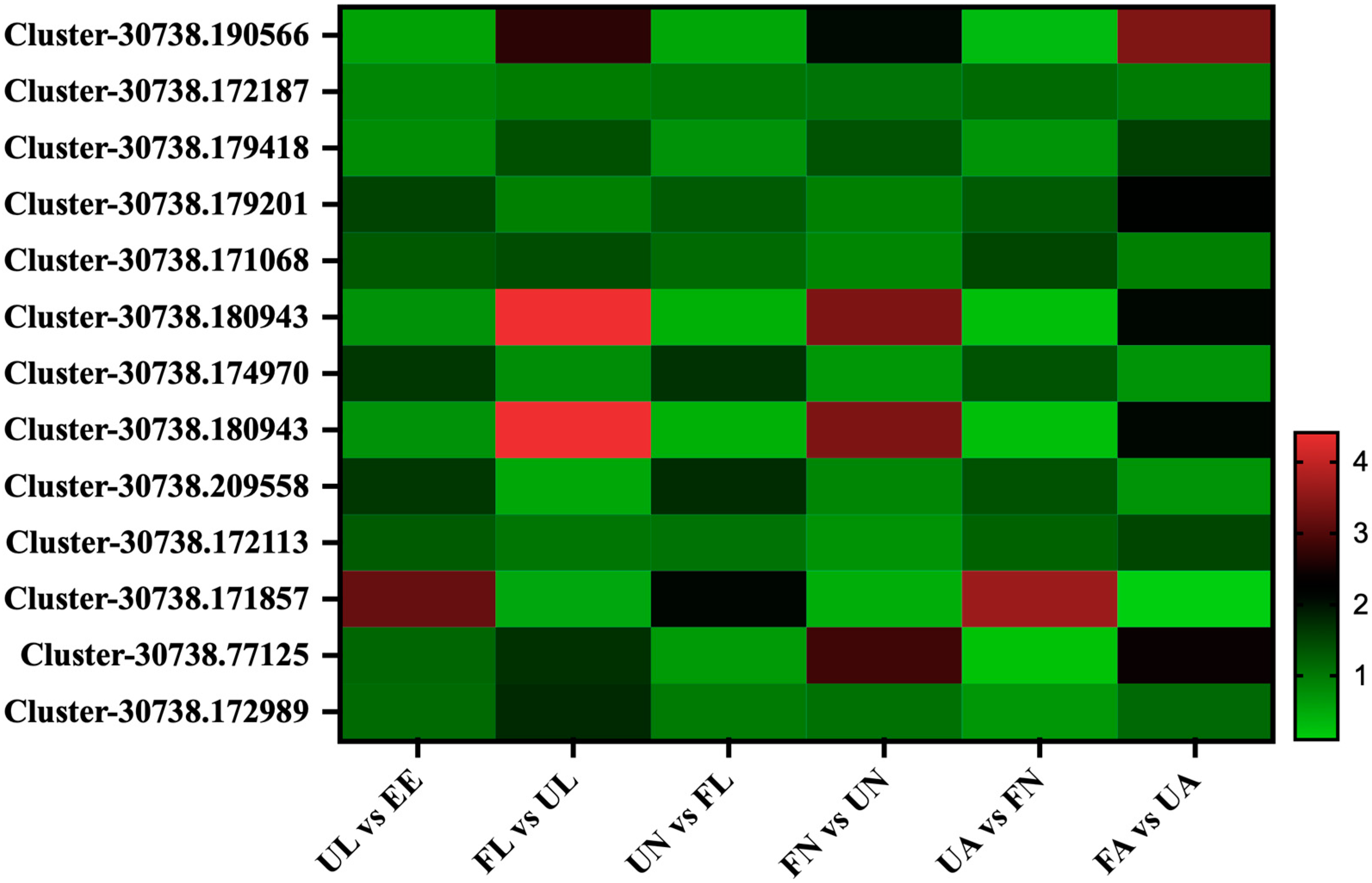
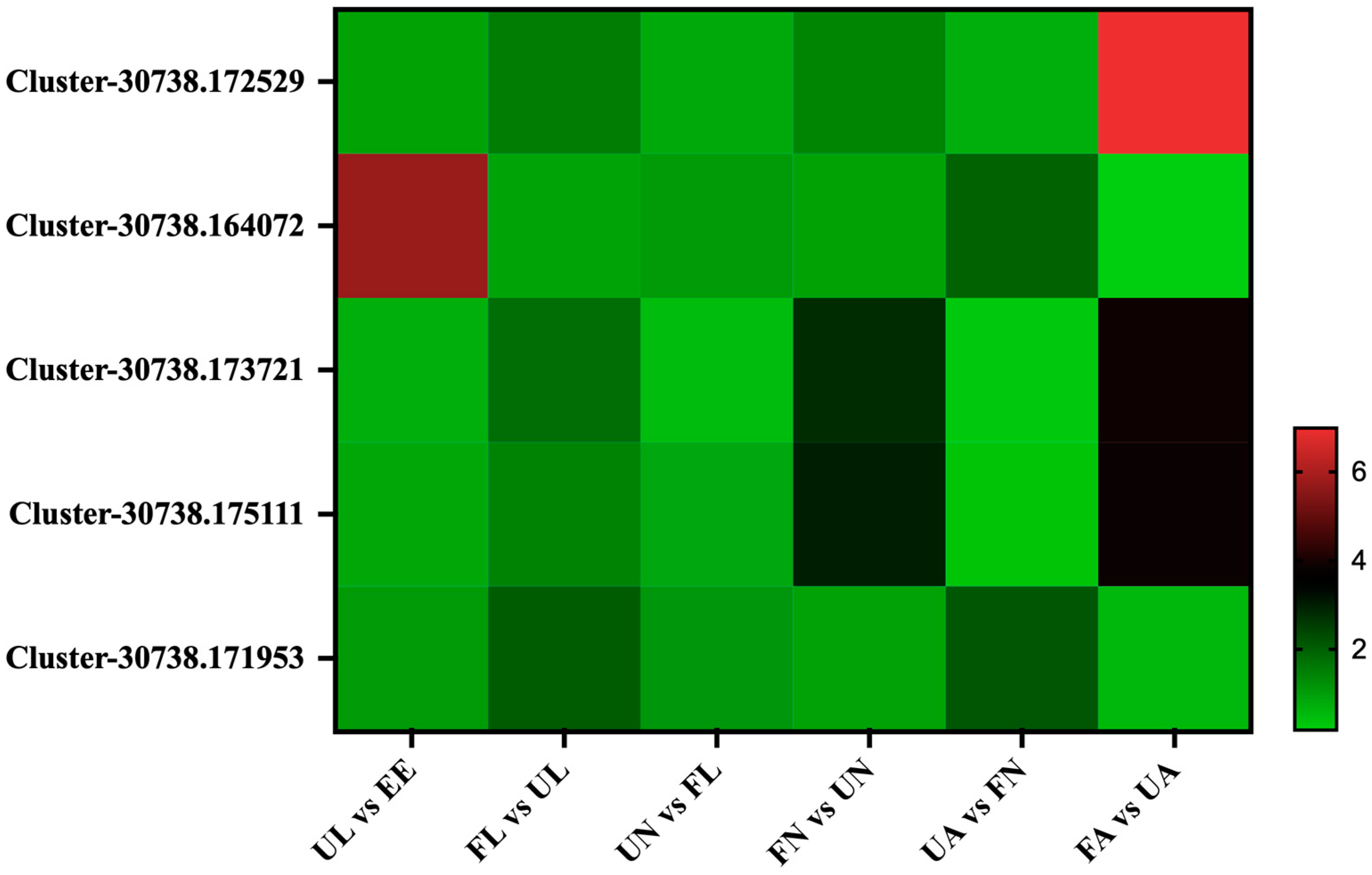
3.4.1. Chitin-Binding Proteins
3.4.2. Digestion-Related Proteins
3.4.3. Vitellogenin Proteins
3.4.4. Cuticle Proteins
3.4.5. Membrane Proteins
3.4.6. Salivary Proteins
3.4.7. Secreted Proteins
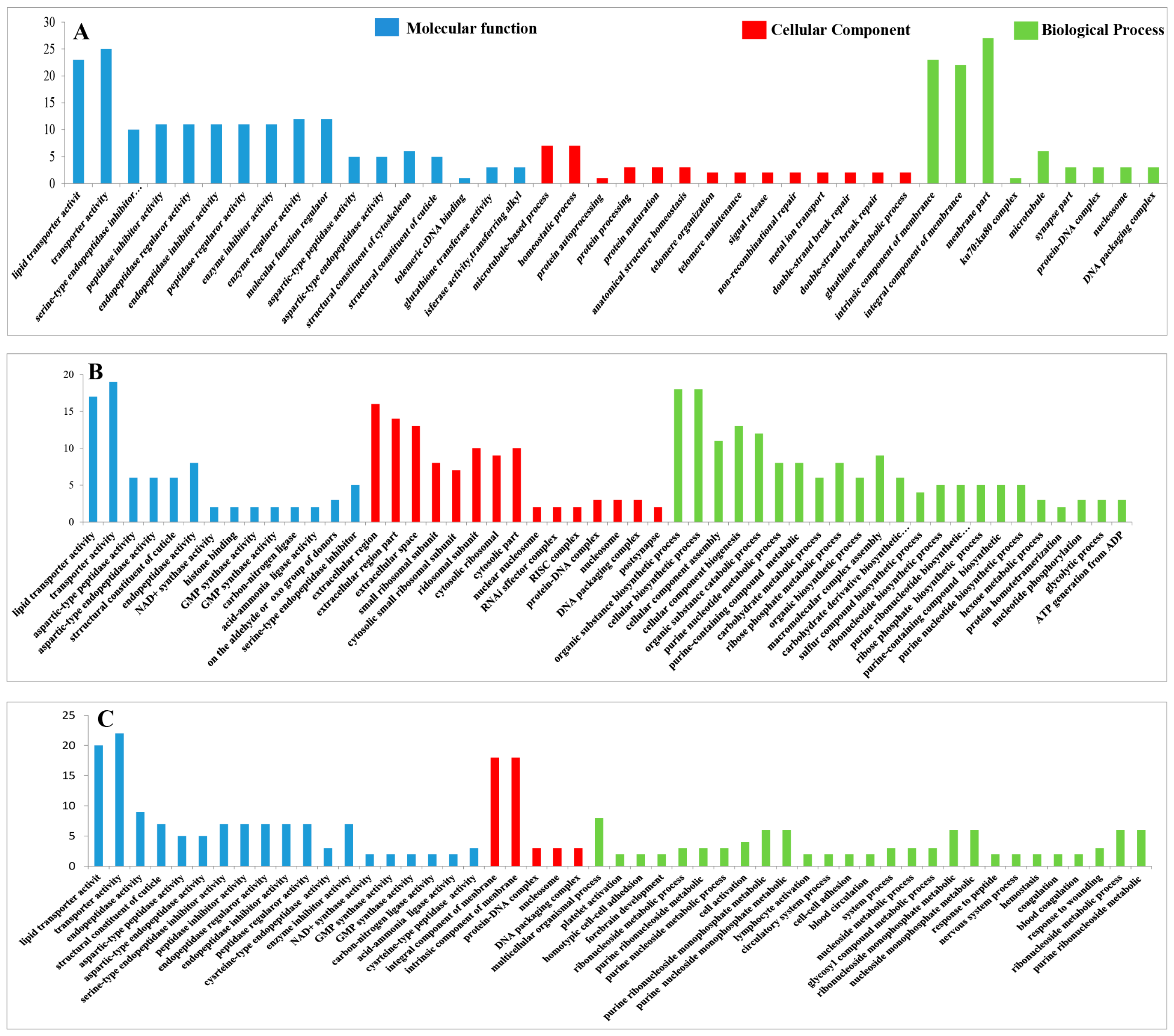
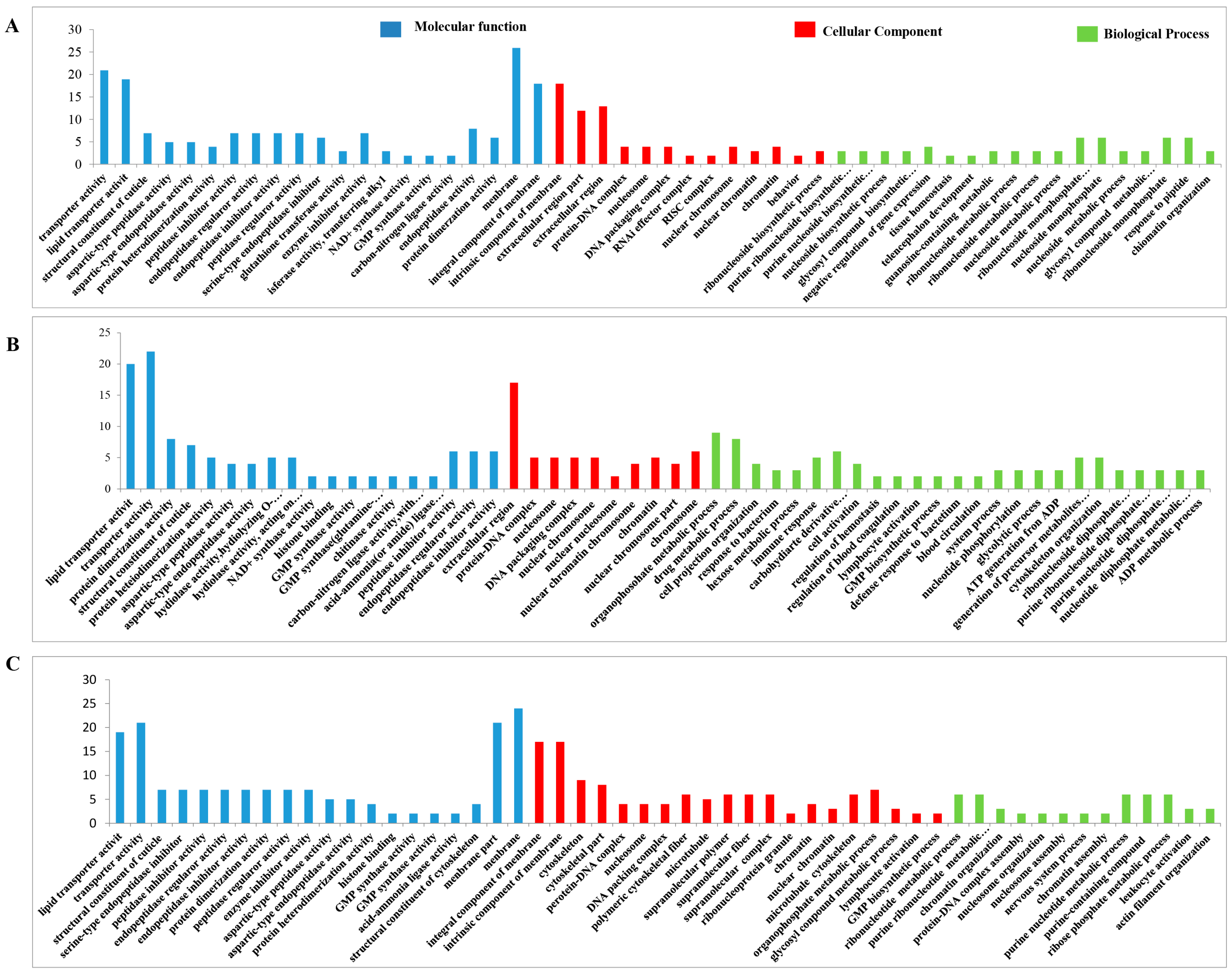
3.4.8. GO Analysis of the DEPs
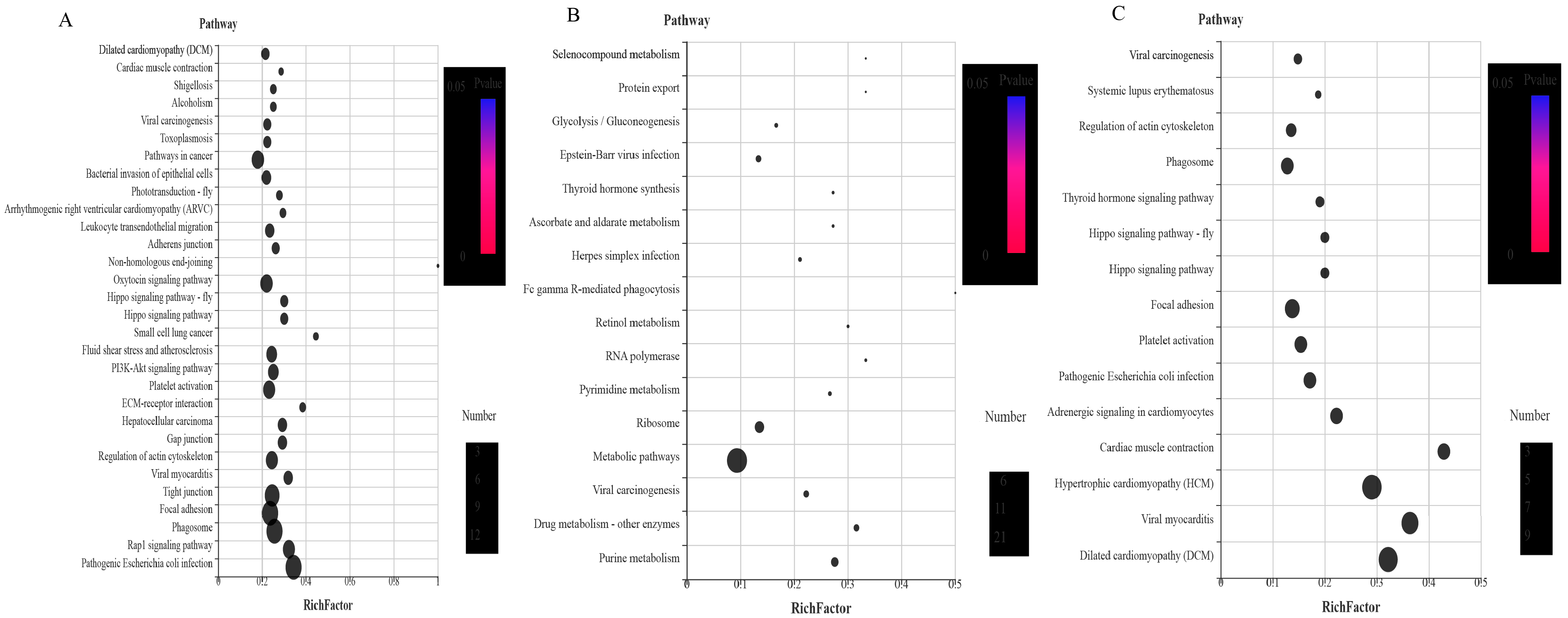
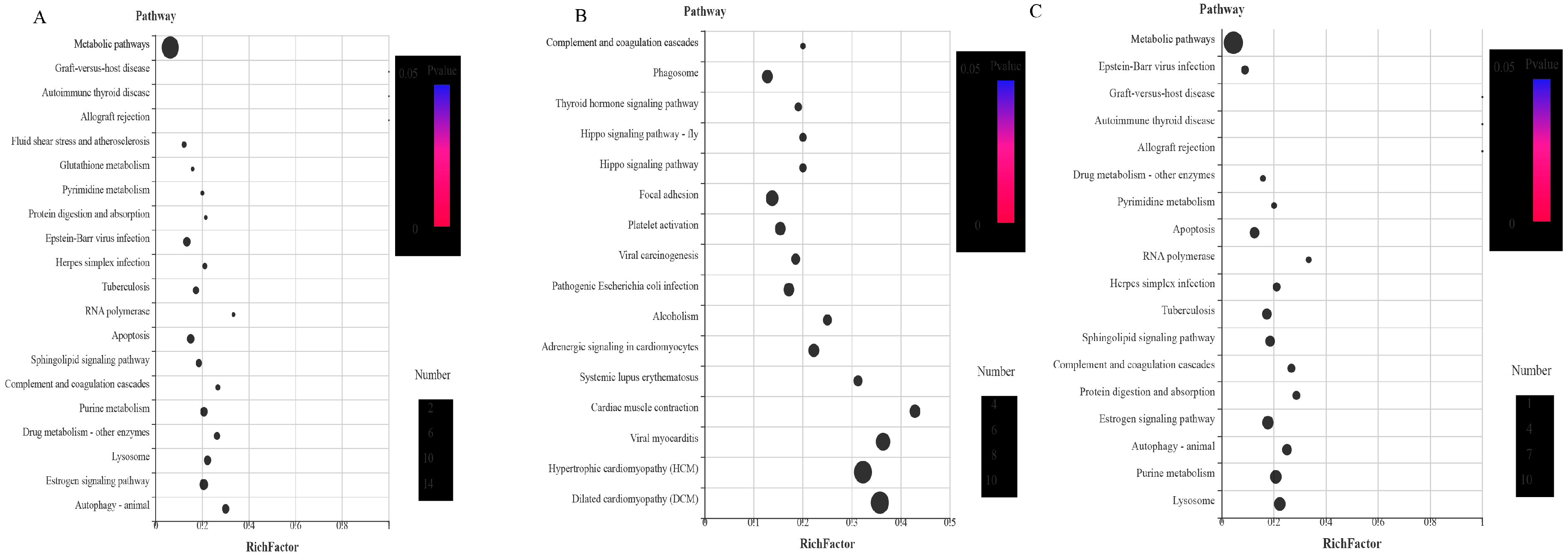
3.4.9. KEGG Analysis of the DEPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abd-Rani, P.A.; Irwin, P.J.; Coleman, G.T.; Gatne, M.; Traub, R.J. A survey of canine tick-borne diseases in India. Parasites Vectors 2011, 4, 141. [Google Scholar] [CrossRef]
- Durden, L.A.; Merker, S.; Beati, L. The tick fauna of Sulawesi, Indonesia (Acari: Ixodoidea: Argasidae and Ixodidae). Exp. Appl. Acarol. 2008, 45, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Wang, J.; Shi, W.; Du, L.; Ye, R.Z.; Zhao, F.; Cao, W.C. Haemaphysalis longicornis. Trends Genet. 2021, 37, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, X.; Bu, F.; Yang, X.; Liu, J. Morphological, biological and molecular characteristics of bisexual and parthenogenetic Haemaphysalis longicornis. Vet. Parasitol. 2012, 189, 344–352. [Google Scholar] [CrossRef]
- Di-Venere, M.; Fumagalli, M.; Cafiso, A.; De-Marco, L.; Epis, S.; Plantard, O.; Bardoni, A.; Salvini, R.; Viglio, S.; Bazzocchi, C.; et al. Ixodes ricinus and Its Endosymbiont Midichloria mitochondrii: A Comparative Proteomic Analysis of Salivary Glands and Ovaries. PLoS ONE 2015, 10, e0138842. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Slam, M.S.; You, M.J. Proteomic screening of antigenic proteins from the hard tick, Haemaphysalis longicornis (Acari: Ixodidae). Korean J. Parasitol. 2015, 53, 85–93. [Google Scholar] [CrossRef]
- Oleaga, A.; Obolo-Mvoulouga, P.; Manzano-Román, R.; Pérez-Sánchez, R. A proteomic insight into the midgut proteome of Ornithodoros moubata females reveals novel information on blood digestion in argasid ticks. Parasites Vectors 2017, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.W.; Wang, G.; Baek, S.J.; Shen, R.F. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel-or LC-MALDI TOF/TOF. J. Proteome Res. 2006, 5, 651–658. [Google Scholar] [CrossRef]
- Pierce, A.; Unwin, R.D.; Evans, C.A.; Griffiths, S.; Carney, L.; Zhang, L.; Jaworska, E.; Lee, C.F.; Blinco, D.; Okoniewski, M.J.; et al. Eight-channel iTRAQ enables comparison of the activity of six leukemogenic tyrosine kinases. Mol. Cell. Proteom. 2008, 7, 853–863. [Google Scholar] [CrossRef]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Wang, X.; Zhang, B.; Wang, M.; Yang, X.; Han, X.; Wang, R.; Ren, S.; Hu, Y.; et al. Comprehensive Analysis of the Global Protein Changes That Occur During Salivary Gland Degeneration in Female Ixodid Ticks Haemaphysalis longicornis. Front. Physiol. 2019, 9, 1943. [Google Scholar] [CrossRef]
- Wang, M.; Hu, Y.; Li, M.; Xu, Q.; Zhang, X.; Wang, X.; Xue, X.; Xiao, Q.; Liu, J.; Wang, H. A proteomics analysis of the ovarian development in females of Haemaphysalis longicornis. Exp. Appl. Acarol. 2020, 80, 289–309. [Google Scholar] [CrossRef]
- Liu, W.G.; Luo, J.; Ren, Q.Y.; Qu, Z.Q.; Lin, H.L.; Xu, X.F.; Ni, J.; Xiao, R.H.; Chen, R.G.; Rashid, M.; et al. A Novel miRNA-hlo-miR-2- Serves as a Regulatory Factor That Controls Molting Events by Targeting CPR1 in Haemaphysalis longicornis Nymphs. Front. Microbiol. 2020, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Oleaga, A.; Obolo-Mvoulouga, P.; Manzano-Román, R.; Pérez-Sánchez, R. Midgut proteome of an argasid tick, Ornithodoros erraticus: A comparison between unfed and engorged females. Parasites Vectors 2015, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Tenzer, S.; Hackenberg, M.; Erhart, J.; Gerhold-Ay, A.; Mazur, J.; Kuharev, J.; Ribeiro, J.M.; Kotsyfakis, M. A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol. Cell. Proteom. 2014, 13, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Heekin, A.M.; Guerrero, F.D.; Bendele, K.G.; Saldivar, L.; Scoles, G.A.; Dowd, S.E.; Gondro, C.; Nene, V.; Djikeng, A.; Brayton, K.A. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol. Res. 2013, 112, 3075–3090. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.L.; Yu, X.B.; Zhang, H.Y.; Wang, H.; Huang, X.Z.; Shen, Y.H.; Lu, C. Identification of Peritrophins and Antiviral Effect of Bm01504 against BmNPV in the Silkworm, Bombyx mori. Int. J. Mol. Sci. 2020, 21, 7973. [Google Scholar] [CrossRef] [PubMed]
- Tellam, R.L.; Eisemann, C. Chitin is only a minor component of the peritrophic matrix from larvae of Lucilia cuprina. Insect Biochem. Mol. Biol. 2000, 30, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Devenport, M.; Jacobs-Lorena, M. The peritrophic matrix of hematophagous insects. Arch. Insect Biochem. Physiol. 2001, 47, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Sato, M.; Inoue, N.; Yokoyama, N.; Taylor, D.; Fujisaki, K. Morphological studies on the extracellular structure of the midgut of a tick, Haemaphysalis longicornis (Acari: Ixodidae). Parasitol. Res. 2003, 90, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kongsuwan, K.; Josh, P.; Zhu, Y.; Pearson, R.; Goug, J.; Colgrave, M.L. Exploring the midgut proteome of partially fed female cattle tick (Rhipicephalus (Boophilus) microplus). J. Insect Physiol. 2010, 56, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Coons, L.B. The role of the fat body, midgut and ovary in vitellogenin production and vitellogenesis in the female tick, Dermacentor variabilis. Int. J. Parasitol. 1992, 22, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Boldbaatar, D.; Umemiya-Shirafuji, R.; Liao, M.; Tanaka, T.; Xuan, X.; Fujisaki, K. Multiple vitellogenins from the Haemaphysalis longicornis tick are crucial for ovarian development. J. Insect Physiol. 2010, 56, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Xavier, M.A.; Tirloni, L.; Pinto, A.F.; Diedrich, J.K.; Yates, J.R.; Mulenga, A.; Logullo, C.; da Silva Vaz, I., Jr.; Seixas, A.; Termignoni, C. A proteomic insight into vitellogenesis during tick ovary maturation. Sci. Rep. 2018, 8, 4698. [Google Scholar] [CrossRef]
- Willis, J.H. Structural cuticular proteins from arthropods: Annotation, nomenclature, and sequence characteristics in the genomics era. Insect Biochem. Mol. Biol. 2010, 40, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, Y.; Pu, F.; Sun, P.; He, F.; Zhang, L.; Li, Y.; Ma, Z.; Wang, H. Understanding Membrane Protein Drug Targets in Computational Perspective. Curr. Drug Targets 2019, 20, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.S.; Sauer, J.R. Tick salivary glands: Function, physiology and future. Parasitology 2004, 129, S67–S81. [Google Scholar] [CrossRef]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.M.; Mans, B.J. TickSialoFam (TSFam): A Database That Helps to Classify Tick Salivary Proteins, a Review on Tick Salivary Protein Function and Evolution, With Considerations on the Tick Sialome Switching Phenomenon. Front. Cell. Infect. Microbiol. 2020, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, J.; Sanchez-Flores, A.; Cantacessi, C.; Harcus, Y.; Pickering, D.; Bouchery, T.; Camberis, M.; Tang, S.C.; Giacomin, P.; Mulvenna, J.; et al. Secreted proteomes of different developmental stages of the gastrointestinal nematode Nippostrongylus brasiliensis. Mol. Cell. Proteom. 2014, 13, 2736–2751. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Name | Primer Sequence (5′–3′) |
|---|---|---|
| Flotillin | Flotillin F | CCATCAAGGACATCAGCGAT |
| Flotillin R | TATGCAGCCTTCTTGAGCTC | |
| Mo-25 | Mo-25 F | GCACACGTTCACCAACAGTA |
| Mo-25 R | ACAAGATCGCAATCCTCCTG | |
| Dystrophin | Dystrophin F | AACGTGCACGTGTCCATGC |
| Dystrophin R | GAGGTTCTGCAGGAGGTTGA | |
| CRK | CRK F | AGAACGAAGTCGGCGTCTTT |
| CRK R | ATGTCGGGGAACATCTGGT | |
| Septin-1 | Septin-1 F | TGTTATCAGCATGGTCCTGA |
| Septin-1 R | AAGCAGCTCAAGGAGTCCGT | |
| Septin-2 | Septin-2 F | AACAGCTTGTCAGCAAATC |
| Septin-2 R | GACGTTGCCCTCTTGCAATT | |
| β-actin | β-actin F | CGTTCCTGGGTATGGAATCG |
| β-actin R | TCCACGTCGCACTTCATGAT |
| Total-Spectra | Spectra | Peptide | Unique-Peptide | Protein-Identified | |
|---|---|---|---|---|---|
| Repeat 1 | 839,950 | 7932 | 2769 | 2663 | 1353 |
| Repeat 2 | 829,948 | 8019 | 2781 | 2673 | 1323 |
| Repeat 3 | 938,964 | 8951 | 2939 | 2844 | 1437 |
| Total | 2,608,862 | 24,902 | 4405 | 4295 | 2044 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.-X.; Xu, X.-P.; Meng, F.-M.; Zhang, B.; Li, W.-G.; Zhang, Y.-Y.; Zen, Q.-Y.; Liu, W.-G. Developmental Proteomics Reveals the Dynamic Expression Profile of Global Proteins of Haemaphysalis longicornis (Parthenogenesis). Life 2025, 15, 59. https://doi.org/10.3390/life15010059
Liu M-X, Xu X-P, Meng F-M, Zhang B, Li W-G, Zhang Y-Y, Zen Q-Y, Liu W-G. Developmental Proteomics Reveals the Dynamic Expression Profile of Global Proteins of Haemaphysalis longicornis (Parthenogenesis). Life. 2025; 15(1):59. https://doi.org/10.3390/life15010059
Chicago/Turabian StyleLiu, Min-Xuan, Xiao-Pei Xu, Fan-Ming Meng, Bing Zhang, Wei-Gang Li, Yuan-Yuan Zhang, Qiao-Ying Zen, and Wen-Ge Liu. 2025. "Developmental Proteomics Reveals the Dynamic Expression Profile of Global Proteins of Haemaphysalis longicornis (Parthenogenesis)" Life 15, no. 1: 59. https://doi.org/10.3390/life15010059
APA StyleLiu, M.-X., Xu, X.-P., Meng, F.-M., Zhang, B., Li, W.-G., Zhang, Y.-Y., Zen, Q.-Y., & Liu, W.-G. (2025). Developmental Proteomics Reveals the Dynamic Expression Profile of Global Proteins of Haemaphysalis longicornis (Parthenogenesis). Life, 15(1), 59. https://doi.org/10.3390/life15010059