
Gender Differences Are a Leading Factor in 5-Year Survival of Patients with Idiopathic Pulmonary Fibrosis over Antifibrotic Therapy Reduction
 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Diagnostic Criteria
2.2. Pharmacological Protocol
- nintedanib:
- -
- Inclusion Criteria: Age ≥ 40 years, IPF diagnosis according to international guidelines, FVC > 50% of predicted, and DLCO > 30%.
- -
- Exclusion Criteria: ALT and/or AST > 1.5× ULN, total bilirubin >1.5× ULN, high risk of bleeding, INR > 2, PT/PTT > 150% of ULN, scheduled major surgery in the next 3 months, and high risk of thrombosis.
- pirfenidone:
- -
- Inclusion Criteria: Age 40–80 years, IPF diagnosed in the past 48 months, FVC ≥ 50% of predicted value, and DLCO ≥ 35% of predicted value.
- -
- Exclusion Criteria: Hypersensitivity to the active substance or any excipients, severe liver function or terminal liver disease, severe renal impairment (Creatinine Clearance < 30 mL/min), or terminal kidney disease requiring dialysis.
2.3. Pulmonary Function Assessments
2.4. Statistical Analysis
3. Results
3.1. Population and Clinical Characteristics
3.2. Pharmacological Therapy and Dose Adjustment
4. Discussion
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Caminati, A.; Meloni, F.; Fujita, M. Editorial: Idiopathic Pulmonary Fibrosis: Epidemiology, Prognosis and Treatment. Front. Med. 2023, 10, 1195263. [Google Scholar] [CrossRef] [PubMed]
- Sesé, L.; Nunes, H.; Cottin, V.; Israel-Biet, D.; Crestani, B.; Guillot-Dudoret, S.; Cadranel, J.; Wallaert, B.; Tazi, A.; Maître, B.; et al. Gender Differences in Idiopathic Pulmonary Fibrosis: Are Men and Women Equal? Front. Med. 2021, 8, 713698. [Google Scholar] [CrossRef]
- Kalafatis, D.; Gao, J.; Pesonen, I.; Carlson, L.; Sköld, C.M.; Ferrara, G. Gender Differences at Presentation of Idiopathic Pulmonary Fibrosis in Sweden. BMC Pulm. Med. 2019, 19, 222. [Google Scholar] [CrossRef]
- Han, M.K.; Murray, S.; Fell, C.D.; Flaherty, K.R.; Toews, G.B.; Myers, J.; Colby, T.V.; Travis, W.D.; Kazerooni, E.A.; Gross, B.H.; et al. Sex Differences in Physiological Progression of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2008, 31, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Assayag, D.; Morisset, J.; Johannson, K.A.; Wells, A.U.; Walsh, S.L.F. Patient Gender Bias on the Diagnosis of Idiopathic Pulmonary Fibrosis. Thorax 2020, 75, 407–412. [Google Scholar] [CrossRef]
- Ruaro, B.; Pozzan, R.; Confalonieri, P.; Tavano, S.; Hughes, M.; Matucci Cerinic, M.; Baratella, E.; Zanatta, E.; Lerda, S.; Geri, P.; et al. Gastroesophageal Reflux Disease in Idiopathic Pulmonary Fibrosis: Viewer or Actor? To Treat or Not to Treat? Pharmaceuticals 2022, 15, 1033. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic Pulmonary Fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Man, R.K.; Gogikar, A.; Nanda, A.; Janga, L.S.N.; Sambe, H.G.; Yasir, M.; Ramphall, S. A Comparison of the Effectiveness of Nintedanib and Pirfenidone in Treating Idiopathic Pulmonary Fibrosis: A Systematic Review. Cureus 2024, 16, e54268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, X.; Zhang, J. Pirfenidone and Nintedanib Attenuate Pulmonary Fibrosis in Mice by Inhibiting the Expression of JAK2. J. Thorac. Dis. 2024, 16, 1128–1140. [Google Scholar] [CrossRef]
- Chianese, M.; Screm, G.; Salton, F.; Confalonieri, P.; Trotta, L.; Barbieri, M.; Ruggero, L.; Mari, M.; Reccardini, N.; Geri, P.; et al. Pirfenidone and Nintedanib in Pulmonary Fibrosis: Lights and Shadows. Pharmaceuticals 2024, 17, 709. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-Blind, Placebo-Controlled Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of Spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.M.; Zheng, J.; et al. Multi-Ethnic Reference Values for Spirometry for the 3-95-Yr Age Range: The Global Lung Function 2012 Equations. Eur Respir J 2012, 40, 1324–1343. [Google Scholar] [CrossRef]
- Stanojevic, S.; Graham, B.L.; Cooper, B.G.; Thompson, B.R.; Carter, K.W.; Francis, R.W.; Hall, G.L.; Global Lung Function Initiative TLCO working group; Global Lung Function Initiative (GLI) TLCO. Official ERS Technical Standards: Global Lung Function Initiative Reference Values for the Carbon Monoxide Transfer Factor for Caucasians. Eur. Respir. J. 2017, 50, 1700010. [Google Scholar] [CrossRef]
- Cerfolio, R.J.; Bryant, A.S. Different Diffusing Capacity of the Lung for Carbon Monoxide as Predictors of Respiratory Morbidity. Ann. Thorac. Surg. 2009, 88, 405–410; discussion 410–411. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS Statement: Guidelines for the Six-Minute Walk Test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef]
- Białas, A.J.; Iwański, M.; Miłkowska-Dymanowska, J.; Pietrzak, M.; Majewski, S.; Górski, P.; Piotrowski, W. The Prognostic Value of Fixed Time and Self-Paced Walking Tests in Patients Diagnosed with Idiopathic Pulmonary Fibrosis. Adv. Respir. Med. 2021, 89, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Sauleda, J.; Núñez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, C.I.; Barbagelata, E.; Gnerre, G.E.P.; Politi, C. Le Malattie Respiratorie Nell’ottica Di Genere. Ital. J. Med. 2019, 7, 1–33. [Google Scholar] [CrossRef]
- Lee, J.S.; Ryu, J.H.; Elicker, B.M.; Lydell, C.P.; Jones, K.D.; Wolters, P.J.; King, T.E.; Collard, H.R. Gastroesophageal Reflux Therapy Is Associated with Longer Survival in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 1390–1394. [Google Scholar] [CrossRef]
- Jo, H.E.; Corte, T.J.; Glaspole, I.; Grainge, C.; Hopkins, P.M.A.; Moodley, Y.; Reynolds, P.N.; Chapman, S.; Walters, E.H.; Zappala, C.; et al. Gastroesophageal Reflux and Antacid Therapy in IPF: Analysis from the Australia IPF Registry. BMC Pulm. Med. 2019, 19, 84. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Behr, J.; Crestani, B.; Stansen, W.; Schlenker-Herceg, R.; Stowasser, S.; Raghu, G. Anti-Acid Therapy in Idiopathic Pulmonary Fibrosis: Insights from the INPULSIS® Trials. Respir. Res. 2018, 19, 167. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Lee, D.H.; Ahn, B.K.; Hwang, J.J.; Yoon, H.; Shin, C.M.; Park, Y.S.; Kim, N. Protective Effect of Proton Pump Inhibitor for Survival in Patients with Gastroesophageal Reflux Disease and Idiopathic Pulmonary Fibrosis. J. Neurogastroenterol. Motil. 2016, 22, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Kawano-Dourado, L.; Glassberg, M.K.; Assayag, D.; Borie, R.; Johannson, K.A. Sex and Gender in Interstitial Lung Diseases. Eur. Respir. Rev. 2021, 30, 210105. [Google Scholar] [CrossRef]
- Kang, J.; Chung, M.P.; Park, M.S.; Oh, I.J.; Lee, H.B.; Kim, Y.W.; Park, J.S.; Uh, S.T.; Kim, Y.S.; Jegal, Y.; et al. Clinical Outcomes of Dose Modification during Pirfenidone Treatment for IPF: A Nationwide Post-Marketing Surveillance Study. Front. Pharmacol. 2022, 13, 1025947. [Google Scholar] [CrossRef]
- Porse, S.; Hoyer, N.; Shaker, S.B. Impact of Reduction in Antifibrotic Treatment on Mortality in Idiopathic Pulmonary Fibrosis. Respir. Med. 2022, 204, 107015. [Google Scholar] [CrossRef]
- Adegunsoye, A.; Kropski, J.A.; Behr, J.; Blackwell, T.S.; Corte, T.J.; Cottin, V.; Glanville, A.R.; Glassberg, M.K.; Griese, M.; Hunninghake, G.M.; et al. Genetics and Genomics of Pulmonary Fibrosis: Charting the Molecular Landscape and Shaping Precision Medicine. Am. J. Respir. Crit. Care Med. 2024, 210, 401–423. [Google Scholar] [CrossRef] [PubMed]
- Tomos, I.; Roussis, I.; Matthaiou, A.M.; Dimakou, K. Molecular and Genetic Biomarkers in Idiopathic Pulmonary Fibrosis: Where Are We Now? Biomedicines 2023, 11, 2796. [Google Scholar] [CrossRef] [PubMed]
- Greene, K.E.; King, T.E.; Kuroki, Y.; Bucher-Bartelson, B.; Hunninghake, G.W.; Newman, L.S.; Nagae, H.; Mason, R.J. Serum Surfactant Proteins-A and -D as Biomarkers in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2002, 19, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.; Salvini, M.; Fui, A.; Cillis, G.; Cameli, P.; Mazzei, M.A.; Fossi, A.; Refini, R.M.; Rottoli, P. Calgranulin B and KL-6 in Bronchoalveolar Lavage of Patients with IPF and NSIP. Inflammation 2019, 42, 463–470. [Google Scholar] [CrossRef]
- Lacedonia, D.; De Pace, C.C.; Rea, G.; Capitelli, L.; Gallo, C.; Scioscia, G.; Tondo, P.; Bocchino, M. Machine Learning and BMI Improve the Prognostic Value of GAP Index in Treated IPF Patients. Bioengineering 2023, 10, 251. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Total | Male | Female | p | |
|---|---|---|---|---|
| N = 164 | N = 131 | N = 33 | ||
| Age at diagnosis (year) | 69.1 ± 7.7 | 69.3 ± 7.4 | 68.5 ± 9.2 | 0.626 |
| Sex (male) | 131 (80%) | |||
| Type of diagnosis | ||||
| Histological diagnosis | 34 (21%) | 22 (17%) | 12 (36%) | 0.013 |
| Radiological diagnosis | 130 (79%) | 109 (83%) | 21 (64%) | 0.013 |
| Risk factors | ||||
| Smoking habits | 107 (65%) | 97 (74%) | 10 (30%) | <0.001 |
| Packs per years | 44.3 ± 30.9 | 45.8 ± 31.0 | 23.9 ± 21.2 | 0.127 |
| IPF familiar | 33 (20%) | 26 (20%) | 7 (21%) | 0.862 |
| Occupational exposure | 36 (22%) | 29 (22%) | 7 (21%) | 0.909 |
| Comorbidities | ||||
| GERD | 33 (20%) | 17 (13%) | 16 (48%) | <0.001 |
| Pulmonary hypertension | 39 (24%) | 34 (26%) | 5 (15%) | 0.195 |
| Pulmonary neoplasm | 3 (2%) | 3 (2%) | 0 (0%) | 0.383 |
| Depression | 8 (5%) | 5 (4%) | 3 (9%) | 0.211 |
| CVD | 95 (58%) | 77 (59%) | 18 (55%) | 0.662 |
| Rheumatological diseases | 17 (10%) | 16 (12%) | 1 (3%) | 0.123 |
| Antifibrotic therapy | ||||
| nintedanib | 73 (45%) | 56 (43%) | 17 (52%) | 0.368 |
| pirfenidone | 91 (55%) | 75 (57%) | 16 (48%) | 0.368 |
| Suspension therapy | 11 (7%) | 7 (5%) | 4 (12%) | 0.166 |
| Reduction in dosage | 37 (23%) | 28 (21%) | 9 (27%) | 0.472 |
| Reduction in dosage nintedanib/pirfenidone | 28/9 | 21/7 | 7/2 | <0.001 |
| Duration of therapy (mo) | 41.9 ± 18.9 | 39.4 ± 19.2 | 51.7 ± 14.6 | 0.001 |
| AE * | 38 (23%) | 31 (24%) | 7 (21%) | 0.767 |
| Pulmonary performance | ||||
| FEV1 (L) | 2.0 ± 0.6 | 2.1 ± 0.5 | 1.6 ± 0.4 | <0.001 |
| FEV1 (% of pred.) | 82.4 ± 19.7 | 79.4 ± 17.5 | 94.8 ± 23.5 | <0.001 |
| FVC (L) | 2.4 ± 0.8 | 2.6 ± 0.7 | 1.8 ± 0.5 | <0.001 |
| FVC (% of pred.) | 77.4 ± 19.4 | 74.9 ± 18.5 | 87.2 ± 20.1 | 0.001 |
| DLCO | 50.5 ± 14.8 | 50.1 ± 15.0 | 52.1 ± 14.1 | 0.514 |
| 6MWT (mt) | 352.0 ± 135.8 | 356.8 ± 136.2 | 327.1 ± 134.2 | 0.361 |
| SpO2 (6MWT start) | 95.7 ± 2.0 | 95.6 ± 2.1 | 96.3 ± 1.7 | 0.171 |
| SpO2 (6MWT end) | 87.3 ± 10.1 | 87.4 ± 6.5 | 86.5 ± 19.8 | 0.698 |
| Survival | ||||
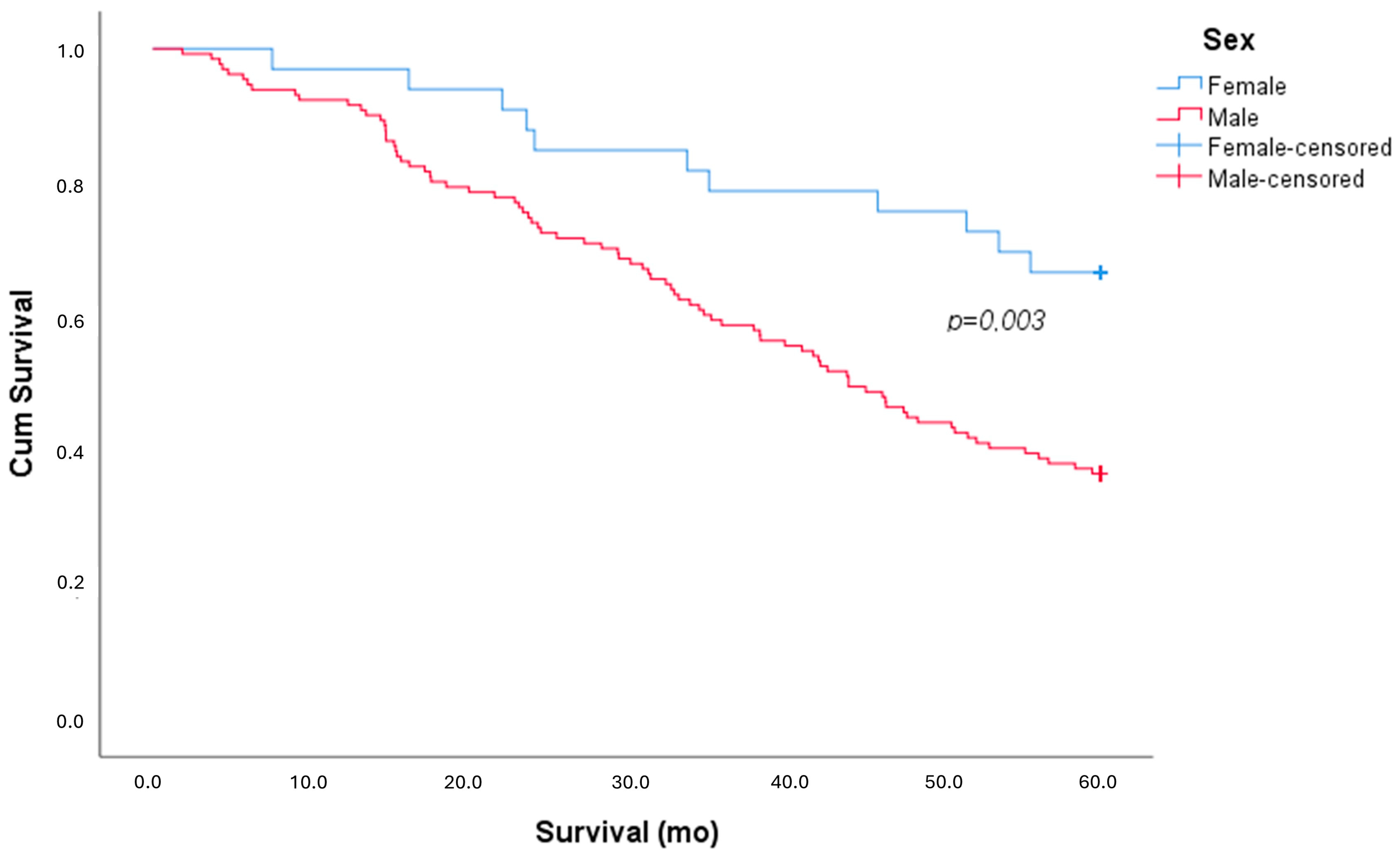
| Exitus | 94 (57%) | 83 (63%) | 11 (33%) | 0.002 |
| Survival (mo) | 42.9 ± 19.0 | 40.8 ± 19.2 | 51.2 ± 15.6 | 0.005 |
| Complete | Reduced | p | |
|---|---|---|---|
| N = 127 | N = 37 | ||
| Age at diagnosis (year) | 69.5 ± 6.9 | 68.0 ± 10.2 | 0.31 |
| Sex (male) | 103 (81%) | 28 (76%) | 0.472 |
| Antifibrotic therapy | |||
| nintedanib/pirfenidone | 62%/90% | 38%/10% | <0.001 |
| Duration of therapy (mo) | 40.5 ± 19.2 | 46.9 ± 17.4 | 0.07 |
| AE * | 26 (20%) | 12 (32%) | 0.131 |
| Pulmonary performance | |||
| FEV1 (L) | 2.0 ± 0.6 | 1.9 ± 0.4 | 0.292 |
| FEV1 (% of pred.) | 83.9 ± 19.9 | 77.3 ± 18.4 | 0.104 |
| FVC (L) | 2.4 ± 0.8 | 2.3 ± 0.7 | 0.532 |
| FVC (% of pred.) | 78.1 ± 19.4 | 75.2 ± 19.4 | 0.423 |
| DLCO | 50.6 ± 14.4 | 50.2 ± 16.1 | 0.896 |
| 6MWT (mt) | 346.2 ± 140.5 | 370.4 ± 119.8 | 0.389 |
| SpO2 (6MWT start) | 95.6 ± 2.1 | 96.3 ± 1.6 | 0.099 |
| SpO2 (6MWT end) | 87.5 ± 6.6 | 86.6 ± 17.2 | 0.699 |
| Survival | |||
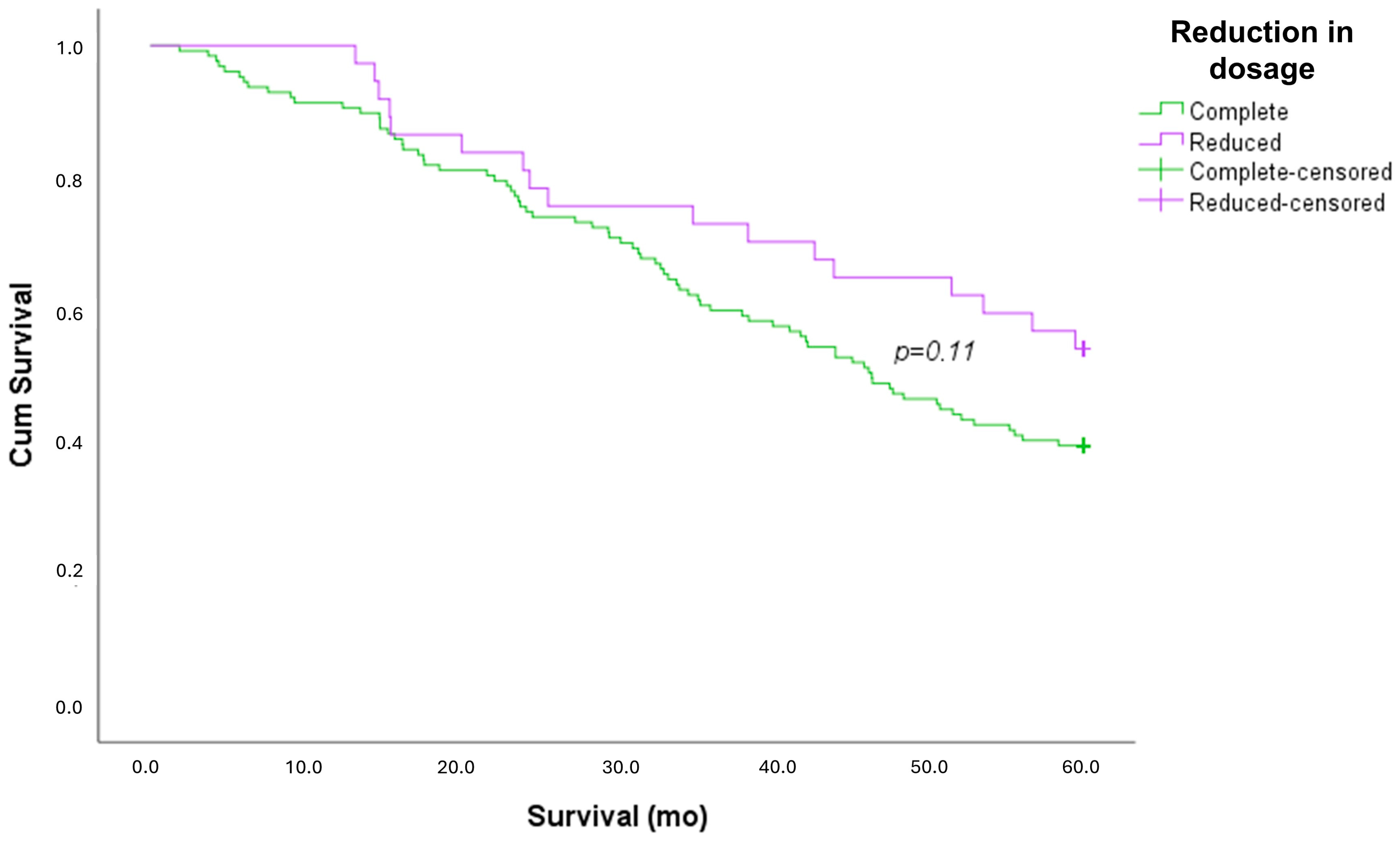
| Exitus | 77 (61%) | 17 (46%) | 0.113 |
| Survival (mo) | 41.6 ± 19.2 | 47.3 ± 17.8 | 0.112 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tondo, P.; Scioscia, G.; De Pace, C.C.; Murgolo, F.; Maci, F.; Stella, G.M.; Pescatore, D.; Foschino Barbaro, M.P.; Lacedonia, D. Gender Differences Are a Leading Factor in 5-Year Survival of Patients with Idiopathic Pulmonary Fibrosis over Antifibrotic Therapy Reduction. Life 2025, 15, 106. https://doi.org/10.3390/life15010106
Tondo P, Scioscia G, De Pace CC, Murgolo F, Maci F, Stella GM, Pescatore D, Foschino Barbaro MP, Lacedonia D. Gender Differences Are a Leading Factor in 5-Year Survival of Patients with Idiopathic Pulmonary Fibrosis over Antifibrotic Therapy Reduction. Life. 2025; 15(1):106. https://doi.org/10.3390/life15010106
Chicago/Turabian StyleTondo, Pasquale, Giulia Scioscia, Cosimo C. De Pace, Fabiola Murgolo, Federica Maci, Giulia M. Stella, Dalila Pescatore, Maria Pia Foschino Barbaro, and Donato Lacedonia. 2025. "Gender Differences Are a Leading Factor in 5-Year Survival of Patients with Idiopathic Pulmonary Fibrosis over Antifibrotic Therapy Reduction" Life 15, no. 1: 106. https://doi.org/10.3390/life15010106
APA StyleTondo, P., Scioscia, G., De Pace, C. C., Murgolo, F., Maci, F., Stella, G. M., Pescatore, D., Foschino Barbaro, M. P., & Lacedonia, D. (2025). Gender Differences Are a Leading Factor in 5-Year Survival of Patients with Idiopathic Pulmonary Fibrosis over Antifibrotic Therapy Reduction. Life, 15(1), 106. https://doi.org/10.3390/life15010106