

Molecular Assessment of Methylglyoxal-Induced Toxicity and Therapeutic Approaches in Various Diseases: Exploring the Interplay with the Glyoxalase System


Abstract
1. Introduction
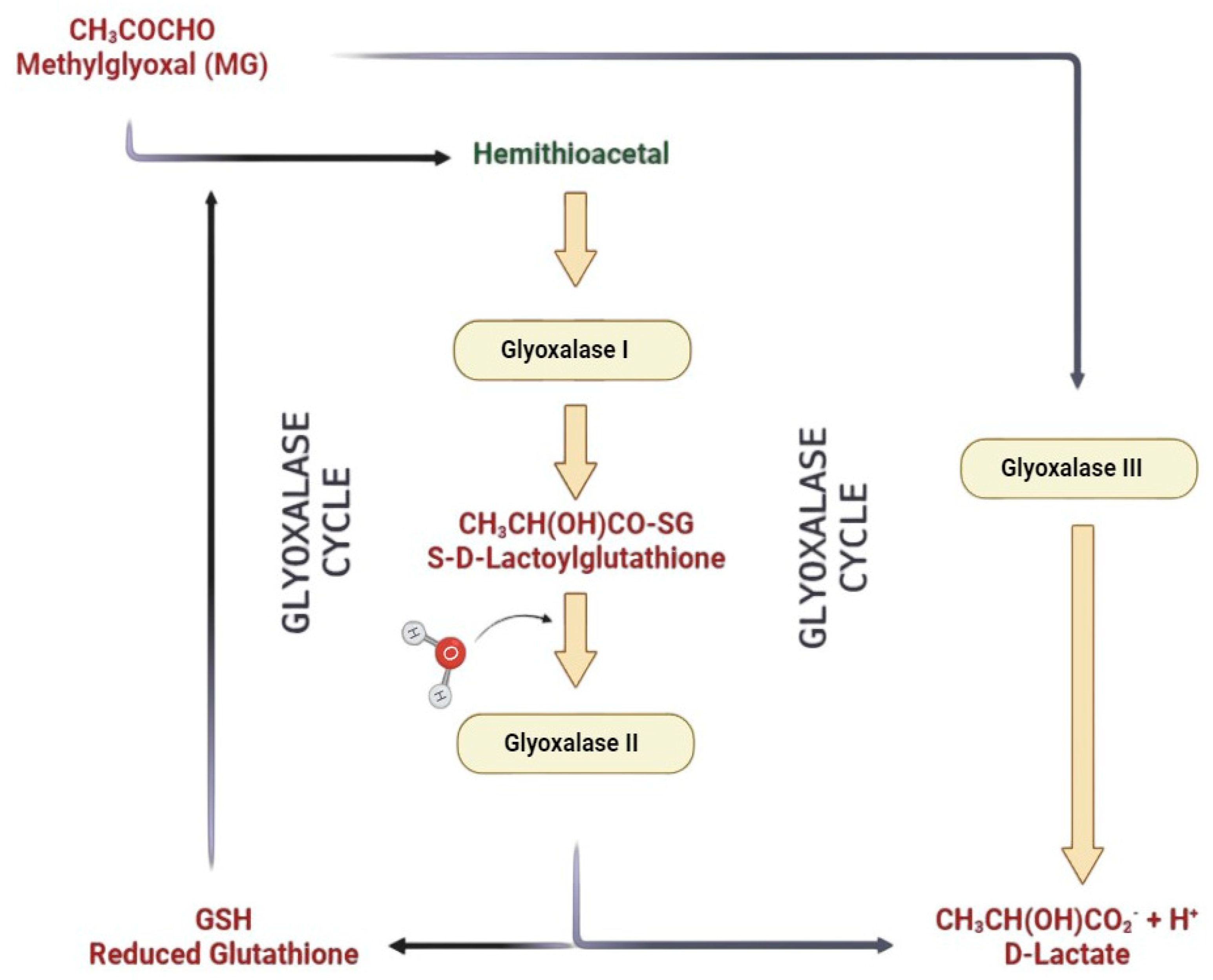
1.1. The Glyoxalase System
1.2. Glyoxalase I
1.3. Glyoxalases II and III
2. MG and GLO in Metabolic Syndrome
2.1. Insulin Resistance (IR) and Diabetes
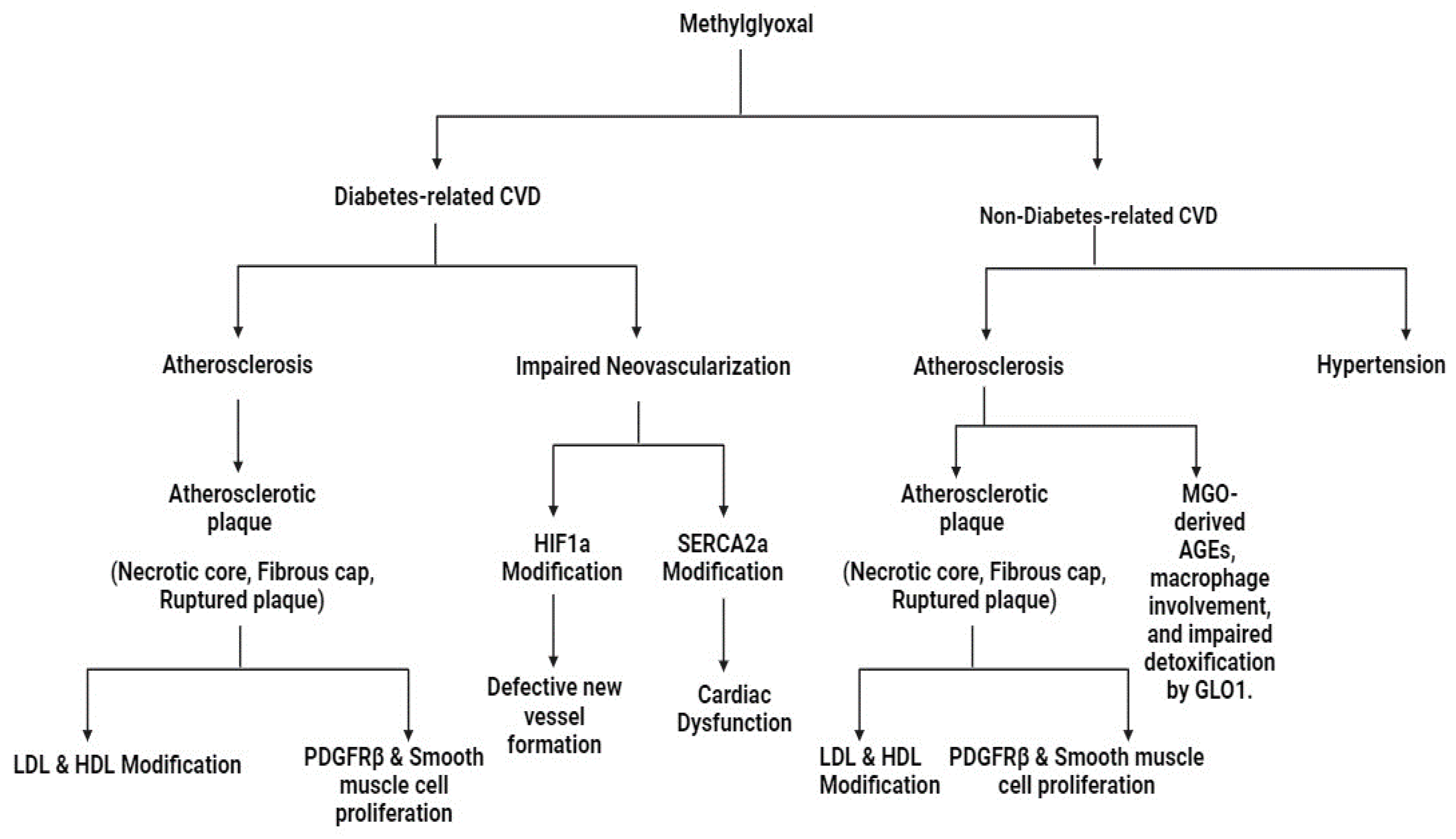
2.2. Cardiovascular Implications
2.3. Obesity and Adipose Tissue Dysfunction
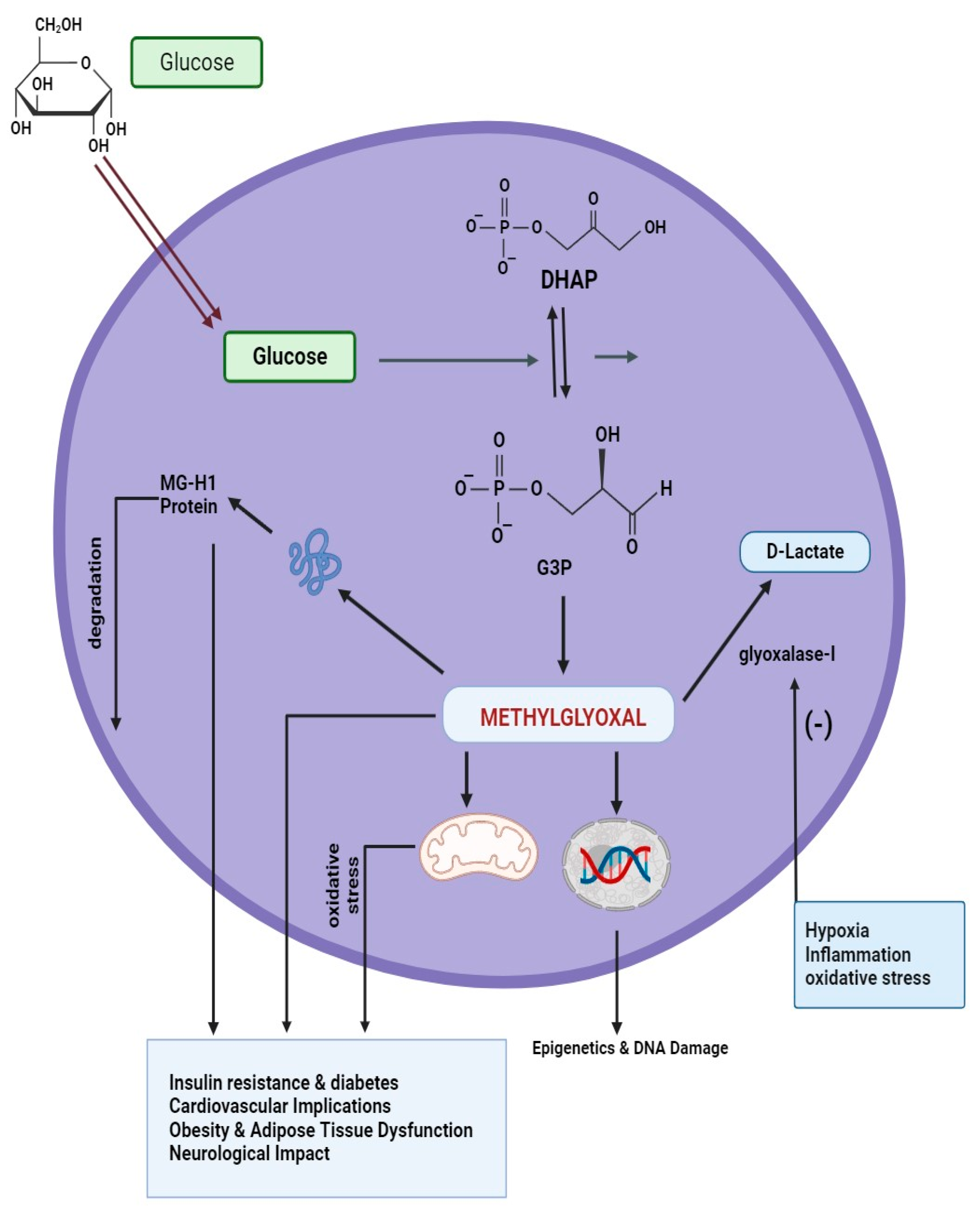
2.4. Oxidative Stress and Inflammation
2.5. Neurological Impact

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Category | Associated Conditions | Role of MG | Role of GLO I | Implications and Findings | Reference |
|---|---|---|---|---|---|
| IR and Diabetes | Elevated MG levels, IR | Exacerbates IR, precursor to type 2 diabetes | Dysregulation leads to MG accumulation, worsening IR | Contributes to diabetes progression, central to metabolic syndrome | [65] |
| Cardiovascular Implications | Increased risk of cardiovascular diseases | Contributes to vascular damage through AGE formation | GLO I mitigates vascular damage by detoxifying MG | Dysfunction in glyoxalase system exacerbates cardiovascular complications associated with metabolic syndrome | [66] |
| Obesity and Adipose Dysfunction | Dysfunctional adipose tissue | Impairs adipocyte function, contributes to obesity-related complications | GLO I regulation of MG levels may influence adipose tissue health | MG and glyoxalase system play roles in adipose tissue dysfunction in metabolic syndrome | [67] |
| Oxidative Stress and Inflammation | Chronic low-grade inflammation, oxidative stress | Pro-oxidant role, contributes to oxidative stress and inflammation | GLO I acts as a defense mechanism, neutralizing MG to prevent damage | Impaired glyoxalase function could worsen oxidative stress and inflammation in metabolic syndrome | [68] |
| Neurological Impact | Implications for neurological health | MG and AGEs induce oxidative stress, contributing to neurodegenerative disorders | GLO I’s role is crucial in mitigating MG-related damage in the brain | Metabolic syndrome may have neurological implications, with MG and GLO I playing key roles in maintaining brain health | [68] |
3. Roles of MG and Glyoxalase in T2D
| Aspect | Key Findings |
|---|---|
| In Vitro Exposure to Elevated Glucose |
|
| Glucose Uptake via GLUT1 |
|
| Experimental Diabetes Studies (Animal Models) |
|
| GLO I Activity in Diabetic Patients |
|
| MG Metabolism Beyond Glyoxalase System |
|
| Pancreatic Beta-Cell Studies |
|
| Protein Modification and AGEs Formation |
|
4. Roles of MG and GLO in T2D Neuropathy
5. Roles of MG and GLO in Stroke
6. Roles of MG and GLO in CVD
6.1. Atherosclerosis
6.2. Hypertension
7. Roles of MG and GLO in Obesity
8. Roles of MG and GLO in Cancer
9. Roles of MG and GLO in Retinopathy
10. Treatment Options with GLO I and MG; Therapeutic GLO 1 Inducers and GLO 1 Inhibitors
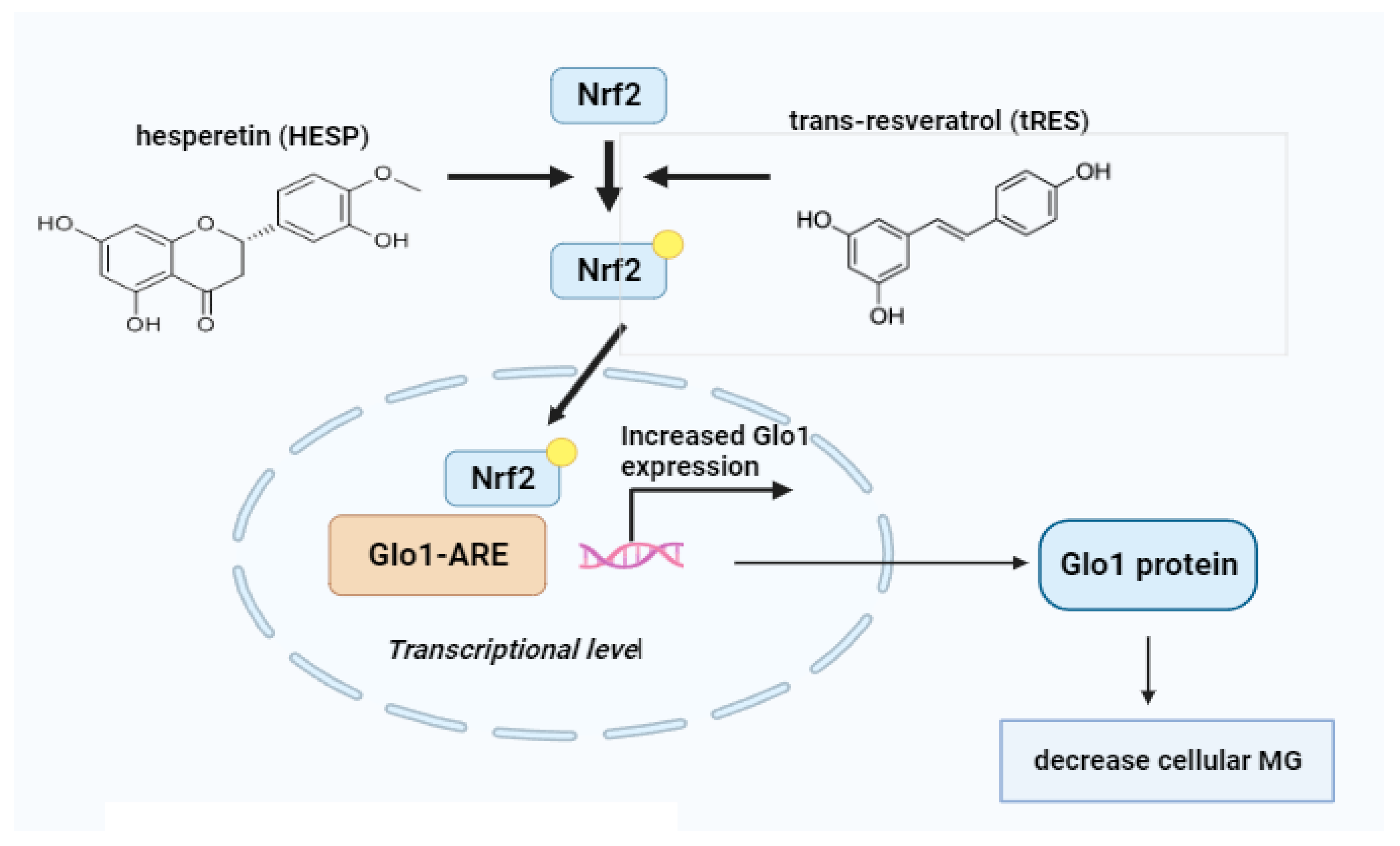
10.1. Glyoxalase 1 Inducers
10.2. Glyoxalase 1 Inhibitors
11. Small-Molecule Regulators of GLO 1
11.1. Small-Molecule Activators of GLO 1
| Small Molecule Activator | Type | Mechanism of Action | Associated Benefits |
|---|---|---|---|
| Candesartan | Synthetic Drug | Restores GLO I function and nitric oxide release affected by angiotensin II | Protective effects in diabetic retinopathy [148]. |
| Resveratrol | Natural Polyphenol | Promotes HO-1 and GLO I expression, inhibits oxidative stress | Counters MG-induced IR, improves metabolic health [149]. |
| Fisetin | Natural Polyphenol | Boosts GLO I activity and expression, increases GSH synthesis | Beneficial for treating diabetic patients, prevents diabetic nephropathy [151]. |
| Mangiferin | Natural C-Glucoside | Enhances GLO I function, inhibits the AGE/RAGE axis and oxidative stress | Prevents diabetic nephropathy [152]. |
| Isothiocyanates | Present in Cruciferous Vegetables | Activates Nrf2-ARE-GLO I pathway | Defends cells against MG and MG-derived AGEs [153] |
| Bardoxolone methyl | Synthetic Compound | Activates Nrf2-Keap1-ARE pathway | Protects function of kidneys in diabetic kidney disease [154]. |
| Pyridoxamine | Vitamin B6 Analogue | Induces expression of GLO I and its activity | Candidate for treating obesity, prevents retinopathy in diabetic rats, quenches MG, increases GLO I activity [155] |
| Aminoguanidine, Alagebrium, Benfotiamine | MG Scavengers | Reduce MG levels, but mechanisms differ from those of GLO I activators | MG scavenging, prevention of MG-induced damage [4] |
11.2. Small-Molecule Inhibitors of GLO
| Inhibitor | Type | Mechanism of Action | Benefits |
|---|---|---|---|
| PBBG | GSH-Based Compound | Potent inhibitory effects on GLO I | Effective GLO I inhibitor, but challenges with cell membrane penetration led to the development of derivatives like BBGC [156]. |
| BBGC | GSH-Based Compound | Efficiently crosses cell membranes, inhibits leukemia cell growth | Improved membrane penetration compared to PBBG, potential for anticancer treatment [156]. |
| S-(N/C-aryl/alkyl-N-hydroxycarbamoyl)glutathione derivatives | GSH-Based Compound | Strong GLO I inhibitory effects, potential selective anticancer agents | Substrates for GLO II, more active in normal cells, offering selectivity for tumor cells [156]. |
| CHG | GSH-Based Compound | Demonstrates competitive inhibition of GLO I, suberate diamide derivatives show stronger effects | Effective competitive inhibitor of GLO I, with potential for enhanced inhibition [156]. |
| Curcumin | Non-GSH Inhibitor | Strong GLO I inhibitory effects | Derived from turmeric, potential anti-tumor agent [157] |
| Flavonoids (myricetin, quercetin, delphinidin) | Non-GSH Inhibitors | Potent GLO I inhibition, hydroxyl group in B ring enhances effectiveness | Natural compounds with potential anticancer properties [158] |
| Methyl-gerfelin | Non-GSH Inhibitor | Competitive GLO I inhibitor | Acts as a competitive inhibitor of GLO I [160] |
| NSAIDs (acemetacin, indomethacin) | Non-GSH Inhibitors | Inhibitory effects on GLO I | Non-steroidal anti-inflammatory drugs show potential as GLO I inhibitors [156] |
| α-Oxo-carbonic acids esters (ethyl pyruvate) | Non-GSH Inhibitor | Exhibits inhibitory effects on GLO I | α-Oxo-carbonic acid esters as potential GLO I inhibitors [156] |
| N-Hydroxypyridones and thiazolyl carboxylic acid derivatives | Various Compounds | Identified potent inhibitors like 4,6-diphenyl-N-hydroxypyridone | Diverse range of compounds explored, contributing to the potential development of antitumor drugs targeting GLO I [156] |
12. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Antognelli, C.; Ferri, I.; Bellezza, G.; Siccu, P.; Love, H.D.; Talesa, V.N.; Sidoni, A. Glyoxalase 2 drives tumorigenesis in human prostate cells in a mechanism involving androgen receptor and p53-p21 axis. Mol. Carcinog. 2017, 56, 2112–2126. [Google Scholar] [CrossRef]
- Silva, M.S.; Gomes, R.A.; Ferreira, A.E.N.; Freire, A.P.; Cordeiro, C. The glyoxalase pathway: The first hundred years… and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Antognelliand, C.; Talesa, V.N. Glyoxalases in Urological Malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities. Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar] [CrossRef]
- Kalapos, M.P.; Antognelli, C.; de Bari, L. Metabolic Shades of S-D-Lactoylglutathione. Antioxidants 2022, 11, 1005. [Google Scholar] [CrossRef]
- Laus, M.N.; Blando, F.; Soccio, M. Glyoxalase I Assay as a Possible Tool for Evaluation of Biological Activity of Antioxidant-Rich Plant Extracts. plants 2023, 12, 1150. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Ma, J.; Zhang, F.; Xu, C. Glyoxalase I in tumor cell proliferation and survival and as a potential target for anticancer therapy. Oncol. Res. Treat. 2014, 37, 570–574. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993, 14, 287–371. [Google Scholar] [CrossRef]
- Clugston, S.L.; Barnard, J.F.J.; Kinach, R.; Miedema, D.; Ruman, R.; Daub, E.; Honek, J.F. Overproduction and characterization of a dimeric non-zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry 1998, 37, 8754–8763. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Activity, regulation, copy number and function in the glyoxalase system. Biochem. Soc. Trans. 2014, 42, 419–424. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef] [PubMed]
- Birkenmeier, G.; Stegemann, C.; Hoffmann, R.; Günther, R.; Huse, K.; Birkemeyer, C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS ONE 2010, 5, e10399. [Google Scholar] [CrossRef] [PubMed]
- De Hemptinne, V.; Rondas, D.; Toepoel, M.; Vancompernolle, K. Phosphorylation on Thr-106 and NO-modification of glyoxalase I suppress the TNF-induced transcriptional activity of NF-kappaB. Mol. Cell. Biochem. 2009, 325, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Van Herreweghe, F.; Mao, J.; Chaplen, F.W.R.; Grooten, J.; Gevaert, K.; Vandekerckhove, J.; Vancompernolle, K. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proc. Natl. Acad. Sci. USA 2002, 99, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Cortizo, F.G.; Pfaff, D.; Wirth, A.; Schlotterer, A.; Medert, R.; Morgenstern, J.; Weber, T.; Hammes, H.-P.; Fleming, T.; Nawroth, P.P.; et al. The activity of glyoxylase 1 is regulated by glucose-responsive phosphorylation on Tyr136. Mol. Metab. 2022, 55, 101406. [Google Scholar] [CrossRef] [PubMed]
- Prevenzano, I.; Leone, A.; Longo, M.; Nicolò, A.; Cabaro, S.; Collina, F.; Panarese, I.; Botti, G.; Formisano, P.; Napoli, R.; et al. Glyoxalase 1 knockdown induces age-related β-cell dysfunction and glucose intolerance in mice. EMBO Rep. 2022, 23, e52990. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Palumbo, I.; Aristei, C.; Talesa, V.N. Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-kappaB. Br. J. Cancer 2014, 111, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Gambelunghe, A.; Talesa, V.N.; Muzi, G. Reactive oxygen species induce apoptosis in bronchial epithelial BEAS-2B cells by inhibiting the antiglycation glyoxalase I defence: Involvement of superoxide anion, hydrogen peroxide and NF-κB. Apoptosis 2014, 19, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Gambelunghe, A.; Del Buono, C.; Murgia, N.; Talesa, V.N.; Muzi, G. Crystalline silica Min-U-Sil 5 induces oxidative stress in human bronchial epithelial cells BEAS-2B by reducing the efficiency of antiglycation and antioxidant enzymatic defenses. Chem. Biol. Interact. 2009, 182, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H(2)O(2) and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef]
- Cordone, V.; Pecorelli, A.; Benedusi, M.; Santini, S.; Falone, S.; Hayek, J.; Amicarelli, F.; Valacchi, G. Antiglycative Activity and RAGE Expression in Rett Syndrome. Cells 2019, 8, 161. [Google Scholar] [CrossRef]
- Cahan, P.; Li, Y.; Izumi, M.; Graubert, T.A. The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat. Genet. 2009, 41, 430–437. [Google Scholar] [CrossRef]
- Marian, A.J. Nature’s genetic gradients and the clinical phenotype. Circ. Cardiovasc. Genet. 2009, 2, 537–539. [Google Scholar] [CrossRef]
- Shafie, A.; Xue, M.; Thornalley, P.J.; Rabbani, N. Copy number variation of glyoxalase I. Biochem. Soc. Trans. 2014, 42, 500–503. [Google Scholar] [CrossRef]
- Talesa, V.N.; Ferri, I.; Bellezza, G.; Love, H.D.; Sidoni, A.; Antognelli, C. Glyoxalase 2 Is Involved in Human Prostate Cancer Progression as Part of a Mechanism Driven By PTEN/PI3K/AKT/mTOR Signaling With Involvement of PKM2 and ERalpha. Prostate 2017, 77, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Du, X.; D’agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Wuand, L.; Juurlink, B.H. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002, 39, 809–814. [Google Scholar]
- Tianand, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 963. [Google Scholar]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief. 2018, 16, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Tatone, C.; Heizenrieder, T.; Di Emidio, G.; Treffon, P.; Amicarelli, F.; Seidel, T.; Eichenlaub-Ritter, U. Evidence that carbonyl stress by methylglyoxal exposure induces DNA damage and spindle aberrations, affects mitochondrial integrity in mammalian oocytes and contributes to oocyte ageing. Hum. Reprod. 2011, 26, 1843–1859. [Google Scholar] [CrossRef]
- Tatone, C.; Eichenlaub-Ritter, U.; Amicarelli, F. Dicarbonyl stress and glyoxalases in ovarian function. Biochem. Soc. Trans. 2014, 42, 433–438. [Google Scholar] [CrossRef]
- Antognelli, C.; Mancuso, F.; Frosini, R.; Arato, I.; Calvitti, M.; Calafiore, R.; Talesa, V.N.; Luca, G. Testosterone and Follicle Stimulating Hormone-Dependent Glyoxalase 1 Up-Regulation Sustains the Viability of Porcine Sertoli Cells through the Control of Hydroimidazolone- and Argpyrimidine-Mediated NF-kappaB Pathway. Am. J. Pathol. 2018, 188, 2553–2563. [Google Scholar] [CrossRef]
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem.-Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Investig. Opthalmology Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef] [PubMed]
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R., Jr.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997, 272, 28218–28226. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Kold-Christensen, R.; Johannsen, M. Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol. Metab. TEM 2020, 31, 81–92. [Google Scholar] [CrossRef]
- Li, D.; Ellis, E.M. Aldo-keto reductase 7A5 (AKR7A5) attenuates oxidative stress and reactive aldehyde toxicity in V79-4 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2014, 28, 707–714. [Google Scholar] [CrossRef]
- Baba, S.P.; Barski, O.A.; Ahmed, Y.; O’Toole, T.E.; Conklin, D.J.; Bhatnagar, A.; Srivastava, S. Reductive metabolism of AGE precursors: A metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes 2009, 58, 2486–2497. [Google Scholar] [CrossRef]
- Jagt, D.L.V.; Hunsaker, L.A. Methylglyoxal metabolism and diabetic complications: Roles of aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and 2-oxoaldehyde dehydrogenase. Chem.-Biol. Interact. 2003, 143–144, 341–351. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight 2019, 4, 12. [Google Scholar] [CrossRef]
- Fujii, E.; Iwase, H.; Ishiikarakasa, I.; Yajima, Y.; Hotta, K. The presence of 2-keto-3-deoxygluconic acid and oxoaldehyde dehydrogenase activity in human erythrocytes. Biochem. Biophys. Res. Commun. 1995, 210, 852–857. [Google Scholar] [CrossRef]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Salomón, T.; Sibbersen, C.; Hansen, J.; Britz, D.; Svart, M.V.; Voss, T.S.; Møller, N.; Gregersen, N.; Jørgensen, K.A.; Palmfeldt, J.; et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chem. Biol. 2017, 24, 935–943.e7. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N. Methylglyoxal and glyoxalase 1—A metabolic stress pathway-linking hyperglycemia to the unfolded protein response and vascular complications of diabetes. Clin. Sci. 2022, 136, 819–824. [Google Scholar] [CrossRef]
- Aragonès, G.; Rowan, S.; Francisco, S.G.; Whitcomb, E.A.; Yang, W.; Perini-Villanueva, G.; Schalkwijk, C.G.; Taylor, A.; Bejarano, E. The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells 2021, 10, 1852. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, D.; Chen, L.; Li, J.; Yuan, G.; Yang, G.; Zhang, H.; Guo, X.; Zhang, J. Glycine increases glyoxalase-1 function by promoting nuclear factor erythroid 2-related factor 2 translocation into the nucleus of kidney cells of streptozotocin-induced diabetic rats. J. Diabetes Investig. 2019, 10, 1189–1198. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front. Cardiovasc. Med. 2022, 9, 959285. [Google Scholar] [CrossRef]
- Hanssen, N.M.; Stehouwer, C.D.; Schalkwijk, C.G. Methylglyoxal and glyoxalase I in atherosclerosis. Biochem. Soc. Trans. 2014, 42, 443–449. [Google Scholar] [CrossRef]
- Dominguez, L.J.; Veronese, N.; Barbagallo, M. Magnesium and Hypertension in Old Age. Nutrients 2020, 13, 139. [Google Scholar] [CrossRef]
- Mutchler, A.P.; Huynh, L.; Patel, R.; Lam, T.; Bain, D.; Jamison, S.; Kirabo, A.; Ray, E.C. The role of dietary magnesium deficiency in inflammatory hypertension. Front. Physiol. 2023, 14, 1167904. [Google Scholar] [CrossRef]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Nuss, P. Anxiety disorders and GABA neurotransmission: A disturbance of modulation. Neuropsychiatr. Dis. Treat. 2015, 11, 165–175. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Jung, J.-H.; Lee, S.-J.; Han, S.-S.; Hong, S.-H. Glyoxalase 1 as a Therapeutic Target in Cancer and Cancer Stem Cells. Mol. Cells 2022, 45, 869–876. [Google Scholar] [CrossRef]
- Vasdev, S.; Stuckless, J. Role of methylglyoxal in essential hypertension. Int. J. Angiol. 2010, 19, e58–e65. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Capó, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic Syndrome is Associated with Oxidative Stress and Proinflammatory State. Antioxidants 2020, 9, 236. [Google Scholar] [CrossRef]
- Price, C.L.; Al Hassi, H.O.S.; English, N.R.; Blakemore, A.I.F.; Stagg, A.J.; Knight, S.C. Methylglyoxal modulates immune responses: Relevance to diabetes. J. Cell. Mol. Med. 2010, 14, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Dunkel, A.; Schmid, C.; Schmitt, S.; Hiltensperger, M.; Lohr, K.; Laketa, V.; Donakonda, S.; Ahting, U.; Lorenz-Depiereux, B.; et al. Regulatory myeloid cells paralyze T cells through cell–cell transfer of the metabolite methylglyoxal. Nat. Immunol. 2020, 21, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.R.; Moinuddin; Habib, S.; Khan, F.; Alam, K.; Ali, A. Structural changes in histone H2A by methylglyoxal generate highly immunogenic amorphous aggregates with implications in auto-immune response in cancer. Glycobiology 2016, 26, 129–141. [Google Scholar] [CrossRef]
- Son, S.; Hwang, I.; Han, S.H.; Shin, J.-S.; Shin, O.S.; Yu, J.-W. Advanced glycation end products impair NLRP3 inflammasome–mediated innate immune responses in macrophages. J. Biol. Chem. 2017, 292, 20437–20448. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-kappaB Pathway. BioMed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Romaldi, B.; Laudadio, E.; Galeazzi, R.; Antognelli, C.; Armeni, T. Glyoxalase 2: Towards a Broader View of the Second Player of the Glyoxalase System. Antioxidants 2022, 11, 2131. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; O’meara, M.; Zhang, X.; Zhang, K.; Seyoum, B.; Yi, Z.; Kaufman, R.J.; Monks, T.J.; Wang, J.-M. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Wasik, A.A.; Lehtonen, S. Glucose Transporters in Diabetic Kidney Disease-Friends or Foes? Front. Endocrinol. 2018, 9, 155. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Newurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Kanwar, M.; Kowluru, R.A. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes 2009, 58, 227–234. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef]
- Šilhavý, J.; Malínská, H.; Hüttl, M.; Marková, I.; Oliyarnyk, O.; Mlejnek, P.; Šimáková, M.; Liška, F.; Kazdová, L.; Moravcová, R.; et al. Downregulation of the Glo1 Gene Is Associated with Reduced Adiposity and Ectopic Fat Accumulation in Spontaneously Hypertensive Rats. Antioxidants 2020, 9, 1179. [Google Scholar] [CrossRef]
- Cunningham, J.; Rodríguez, M.; Messa, P. Magnesium in chronic kidney disease Stages 3 and 4 and in dialysis patients. Clin. Kidney J. 2012, 5, i39–i51. [Google Scholar] [CrossRef]
- Aragonès, G.; Rowan, S.; Francisco, S.G.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants 2020, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 2008, 40, 553–624. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.J.; Davies, J.; Yates, A.P.; Elliott, A.C.; Lovell, J.; Joule, J.A.; Pemberton, P.; Thornalley, P.J.; Best, L. Effects of methylglyoxal on rat pancreatic beta-cells. Biochem. Pharmacol. 1998, 55, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Hong, C.O.; Ha, S.K.; Lee, K.W. Chebulic Acid Prevents Methylglyoxal-Induced Mitochondrial Dysfunction in INS-1 Pancreatic beta-Cells. Antioxidants 2020, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metab. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Ban, I.; Sugawa, H.; Nagai, R. Protein Modification with Ribose Generates N(delta)-(5-hydro-5-methyl-4-imidazolone-2-yl)-ornithine. Int. J. Mol. Sci. 2022, 23, 1224. [Google Scholar] [CrossRef] [PubMed]
- Meireles, P.; Sales-Dias, J.; Andrade, C.M.; Mello-Vieira, J.; Mancio-Silva, L.; Simas, J.P.; Staines, H.M.; Prudencio, M. GLUT1-mediated glucose uptake plays a crucial role during Plasmodium hepatic infection. Cell Microbiol. 2017, 19, e12646. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef]
- Bo, J.; Xie, S.; Guo, Y.; Zhang, C.; Guan, Y.; Li, C.; Lu, J.; Meng, Q.H. Methylglyoxal Impairs Insulin Secretion of Pancreatic beta-Cells through Increased Production of ROS and Mitochondrial Dysfunction Mediated by Upregulation of UCP2 and MAPKs. J. Diabetes Res. 2016, 2016, 2029854. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 42. [Google Scholar] [CrossRef]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.; Jaiswal, M.; Martin, C.; Pop-Busui, R. Glucose control and diabetic neuropathy: Lessons from recent large clinical trials. Curr. Diabetes Rep. 2014, 14, 528. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Mizukami, H.; Sugimoto, K. Mechanism of diabetic neuropathy: Where are we now and where to go? J. Diabetes Investig. 2011, 2, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Barragán-Iglesias, P.; Kuhn, J.; Vidal-Cantú, G.C.; Salinas-Abarca, A.B.; Granados-Soto, V.; Dussor, G.O.; Campbell, Z.T.; Price, T.J. Activation of the integrated stress response in nociceptors drives methylglyoxal-induced pain. Pain 2019, 160, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Coppey, L.J.; Gellett, J.S.; Davidson, E.P.; Dunlap, J.A.; Lund, D.D.; Yorek, M.A. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 2001, 50, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Westerink, J.; Scheijen, J.L.; van der Graaf, Y.; Stehouwer, C.D.; Schalkwijk, C.G.; Algra, A.; Grobbee, R.D.; Rutten, G.E.; Visseren, F.L.; et al. Higher Plasma Methylglyoxal Levels Are Associated with Incident Cardiovascular Disease and Mortality in Individuals With Type 2 Diabetes. Diabetes Care 2018, 41, 1689–1695. [Google Scholar] [CrossRef]
- Hanssen, N.M.; Teraa, M.; Scheijen, J.L.; Van de Waarenburg, M.; Gremmels, H.; Stehouwer, C.D.; Verhaar, M.C.; Schalkwijk, C.G. Plasma Methylglyoxal Levels Are Associated with Amputations and Mortality in Severe Limb Ischemia Patients with and without Diabetes. Diabetes Care 2021, 44, 157–163. [Google Scholar] [CrossRef]
- Schalkwijk, C.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Al-Saoudi, E.; Christensen, M.M.B.; Nawroth, P.; Fleming, T.; Hommel, E.E.; Jørgensen, M.E.; Fleischer, J.; Hansen, C.S. Advanced glycation end-products are associated with diabetic neuropathy in young adults with type 1 diabetes. Front. Endocrinol. 2022, 13, 891442. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Emerging Glycation-Based Therapeutics-Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. Int. J. Mol. Sci. 2022, 23, 2453. [Google Scholar] [CrossRef]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, W.; Lu, H.; Cai, S. Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules 2022, 27, 7905. [Google Scholar] [CrossRef]
- Ciancarelli, I.; Morone, G.; Iosa, M.; Cerasa, A.; Calabrò, R.S.; Iolascon, G.; Gimigliano, F.; Tonin, P.; Ciancarelli, M.G.T. Influence of Oxidative Stress and Inflammation on Nutritional Status and Neural Plasticity: New Perspectives on Post-Stroke Neurorehabilitative Outcome. Nutrients 2022, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, J.R.; Narayanasamy, P. Neuroprotection through flavonoid: Enhancement of the glyoxalase pathway. Redox Biol. 2018, 14, 465–473. [Google Scholar] [CrossRef]
- Schmoch, T.; Uhle, F.; Siegler, B.H.; Fleming, T.; Morgenstern, J.; Nawroth, P.P.; Weigand, M.A.; Brenner, T. The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology. Int. J. Mol. Sci. 2017, 18, 657. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, e126154. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Micali, L.R.; Wouters, K. Advanced glycation endproducts in diabetes-related macrovascular complications: Focus on methylglyoxal. Trends Endocrinol. Metab. 2023, 34, 49–60. [Google Scholar] [CrossRef]
- Memon, M.; Khan, R.; Riaz, S.; Ain, Q.; Ahmed, M.; Kumar, N. Methylglyoxal and insulin resistance in berberine-treated type 2 diabetic patients. J. Res. Med. Sci. 2018, 23, 110. [Google Scholar] [CrossRef]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of LDL by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Scheijen, J.L.; Stehouwer, C.D.; Wouters, K.; Schalkwijk, C.G. Increased methylglyoxal formation in plasma and tissues during a glucose tolerance test is derived from exogenous glucose. Clin. Sci. 2023, 137, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Bobryshev, Y.V.; Sobenin, I.A.; Melnichenko, A.A.; Chistiakov, D.A. Modified low density lipoprotein and lipoprotein-containing circulating immune complexes as diagnostic and prognostic biomarkers of atherosclerosis and type 1 diabetes macrovascular disease. Int. J. Mol. Sci. 2014, 15, 12807–12841. [Google Scholar] [CrossRef]
- Dhar, I.; Dhar, A.; Wu, L.; Desai, K.M. Methylglyoxal, a reactive glucose metabolite, increases renin angiotensin aldosterone and blood pressure in male Sprague-Dawley rats. Am. J. Hypertens. 2014, 27, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Misko, T.P.; Lin, R.F.; Hickey, W.F.; Trotter, J.L.; Tilton, R.G. Aminoguanidine, an inhibitor of inducible nitric oxide synthase, ameliorates experimental autoimmune encephalomyelitis in SJL mice. J. Clin. Investig. 1994, 93, 2684–2690. [Google Scholar] [CrossRef]
- Fruh, S.M. Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. J. Am. Assoc. Nurse Pract. 2017, 29, S3–S14. [Google Scholar] [CrossRef]
- Haslam, D.W.; James, W.P. Obesity. Lancet 2005, 366, 1197–1209. [Google Scholar] [CrossRef]
- Jia, X.; Chang, T.; Wilson, T.W.; Wu, L. Methylglyoxal mediates adipocyte proliferation by increasing phosphorylation of Akt1. PLoS ONE 2012, 7, e36610. [Google Scholar] [CrossRef]
- Rodrigues, L.; Matafome, P.; Crisóstomo, J.; Santos-Silva, D.; Sena, C.; Pereira, P.; Seiça, R. Advanced glycation end products and diabetic nephropathy: A comparative study using diabetic and normal rats with methylglyoxal-induced glycation. J. Physiol. Biochem. 2014, 70, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Crisóstomo, J.; Matafome, P.; Santos-Silva, D.; Rodrigues, L.; Sena, C.; Pereira, P.; Seiça, R. Methylglyoxal chronic administration promotes diabetes-like cardiac ischaemia disease in Wistar normal rats. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Matafome, P.; Seica, R. Methylglyoxal further impairs adipose tissue metabolism after partial decrease of blood supply. Arch. Physiol. Biochem. 2013, 119, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.T.; Merchant, M.L.; Kain, A.B.; Jevans, A.W.; McLeish, K.R.; Klein, J.B. Proteomic analysis defines altered cellular redox pathways and advanced glycation end-product metabolism in glomeruli of db/db diabetic mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F1157–F1165. [Google Scholar] [CrossRef] [PubMed]
- Atkins, T.W.; Thornally, P.J. Erythrocyte glyoxalase activity in genetically obese (ob/ob) and streptozotocin diabetic mice. Diabetes Res. 1989, 11, 125–129. [Google Scholar]
- Kim, D.H.; Joo, J.I.; Choi, J.-W.; Yun, J.W. Differential expression of skeletal muscle proteins in high-fat diet-fed rats in response to capsaicin feeding. Proteomics 2010, 10, 2870–2881. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 2011, 22, 309–317. [Google Scholar] [CrossRef]
- Apple, M.A.; Greenberg, D.M. Arrest of cancer in mice by therapy with normal metabolites. II. Indefinite survirors among mice treated with mixtures of 2-oxopropanal (NSC-79019) and 2,3-dihydroxypropanal (NSC67934). Cancer Chemother. Rep. 1968, 52, 687–696. [Google Scholar]
- Ayoub, F.M.; Allen, R.E.; Thornalley, P.J. Inhibition of proliferation of human leukaemia 60 cells by methylglyoxal in vitro. Leuk. Res. 1993, 17, 397–401. [Google Scholar] [CrossRef]
- Ray, M.; Halder, J.; Dutta, S.K.; Ray, S. Inhibition of respiration of tumor cells by methylglyoxal and protection of inhibition by lactaldehyde. Int. J. Cancer 1991, 47, 603–609. [Google Scholar] [CrossRef]
- Milanesa, D.M.; Choudhury, M.S.; Mallouh, C.; Tazaki, H.; Konno, S. Methylglyoxal-induced apoptosis in human prostate carcinoma: Potential modality for prostate cancer treatment. Eur. Urol. 2000, 37, 728–734. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Yang, X.; Lu, P.; Yan, X.; Xiao, F.; Zhou, H.; Wen, C.; Shi, M.; Lu, J.; et al. Effects of methylglyoxal and glyoxalase I inhibition on breast cancer cells proliferation, invasion, and apoptosis through modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol. Ther. 2016, 17, 169–180. [Google Scholar] [CrossRef]
- Loarca, L.; Sassi-Gaha, S.; Artlett, C.M. Two alpha-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a p53-dependent manner. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2013, 45, 938–946. [Google Scholar]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Katsube, Y.; Iwaoki, Y.; Silverberg, S.G.; Fujiwara, A. Sclerosing stromal tumor of the ovary associated with endometrial adenocarcinoma: A case report. Gynecol. Oncol. 1988, 29, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.G.; Smith, D.G.; Bhat, M.; Nagaraj, R.H. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J. Biol. Chem. 2006, 281, 11864–11871. [Google Scholar] [CrossRef] [PubMed]
- Berner, A.K.; Brouwers, O.; Pringle, R.; Klaassen, I.; Colhoun, L.; McVicar, C.; Brockbank, S.; Curry, J.W.; Miyata, T.; Brownlee, M.; et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia 2012, 55, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.; Campos, M.C.; Nawroth, P.; Fleming, T. The Glyoxalase System—New Insights into an Ancient Metabolism. Antioxidants 2020, 9, 939. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Gonzalez, E.D.J.L.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.-B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- McMurray, K.M.; Distler, M.G.; Sidhu, P.S.; Cook, J.M.; Arnold, L.A.; Palmer, A.A.; Plant, L.D. Glo1 inhibitors for neuropsychiatric and anti-epileptic drug development. Biochem. Soc. Trans. 2014, 42, 461–467. [Google Scholar] [CrossRef]
- Tamori, S.; Nozaki, Y.; Motomura, H.; Nakane, H.; Katayama, R.; Onaga, C.; Kikuchi, E.; Shimada, N.; Suzuki, Y.; Noike, M.; et al. Glyoxalase 1 gene is highly expressed in basal-like human breast cancers and contributes to survival of ALDH1-positive breast cancer stem cells. Oncotarget 2018, 9, 36515–36529. [Google Scholar] [CrossRef]
- Stratmann, B. Dicarbonyl Stress in Diabetic Vascular Disease. Int. J. Mol. Sci. 2022, 23, 6186. [Google Scholar] [CrossRef]
- Mawson, A.R. The pathogenesis of malaria: A new perspective. Ann. Trop. Med. Parasitol. 2013, 107, 122–129. [Google Scholar] [CrossRef]
- Distler, M.G.; Palmer, A.A. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: Recent advances and mechanistic insights. Front. Genet. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.G.; Tan, G.; Binger, K.J.; Pickering, R.J.; Thomas, M.C.; Nagaraj, R.H.; Cooper, M.E.; Wilkinson-Berka, J.L. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes 2010, 59, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-S.; Cheng, Y.-H.; Chiou, C.-H.; Chang, T.-L. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J. Agric. Food Chem. 2012, 60, 9180–9187. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Inagi, R. Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis. Int. J. Mol. Sci. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef]
- Imran, M.; Arshad, M.S.; Butt, M.S.; Kwon, J.-H.; Sultan, M.T. Mangiferin: A natural miracle bioactive compound against lifestyle related disorders. Lipids Heal. Dis. 2017, 16, 84. [Google Scholar] [CrossRef]
- Schalkwijk, C.G. Vascular AGE-ing by methylglyoxal: The past, the present and the future. Diabetologia 2015, 58, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Isac, P.N.S.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative Stress and NRF2/KEAP1/ARE Pathway in Diabetic Kidney Disease (DKD): New Perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef]
- Stitt, A.; Gardiner, T.A.; Anderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef]
- Santel, T.; Pflug, G.; Hemdan, N.Y.A.; Schäfer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Al-Balas, Q.; Hassan, M.; Al-Oudat, B.; Alzoubi, H.; Mhaidat, N.; Almaaytah, A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules 2012, 17, 13740–13758. [Google Scholar] [CrossRef]
- Kawatani, M.; Okumura, H.; Honda, K.; Kanoh, N.; Muroi, M.; Dohmae, N.; Takami, M.; Kitagawa, M.; Futamura, Y.; Imoto, M.; et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc. Natl. Acad. Sci. USA 2008, 105, 11691–11696. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.S.; Wortmann, M.; Fleming, T.H.; Nawroth, P.P.; Bruckner, T.; Böckler, D.; Hakimi, M. Effect of metformin treatment in patients with type 2 diabetes with respect to glyoxalase 1 activity in atherosclerotic lesions. Vasa 2019, 48, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current knowledge. J. Res. Med. Sci. 2014, 19, 658–664. [Google Scholar] [PubMed]
- Mu, N.; Xu, T.; Gao, M.; Dong, M.; Tang, Q.; Hao, L.; Wang, G.; Li, Z.; Wang, W.; Yang, Y.; et al. Therapeutic effect of metformin in the treatment of endometrial cancer. Oncol. Lett. 2020, 20, 156. [Google Scholar] [CrossRef]
- Antognelli, C.; Cecchetti, R.; Riuzzi, F.; Peirce, M.J.; Talesa, V.N. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J. Cell Mol. Med. 2018, 22, 2865–2883. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhujaily, M. Molecular Assessment of Methylglyoxal-Induced Toxicity and Therapeutic Approaches in Various Diseases: Exploring the Interplay with the Glyoxalase System. Life 2024, 14, 263. https://doi.org/10.3390/life14020263
Alhujaily M. Molecular Assessment of Methylglyoxal-Induced Toxicity and Therapeutic Approaches in Various Diseases: Exploring the Interplay with the Glyoxalase System. Life. 2024; 14(2):263. https://doi.org/10.3390/life14020263
Chicago/Turabian StyleAlhujaily, Muhanad. 2024. "Molecular Assessment of Methylglyoxal-Induced Toxicity and Therapeutic Approaches in Various Diseases: Exploring the Interplay with the Glyoxalase System" Life 14, no. 2: 263. https://doi.org/10.3390/life14020263
APA StyleAlhujaily, M. (2024). Molecular Assessment of Methylglyoxal-Induced Toxicity and Therapeutic Approaches in Various Diseases: Exploring the Interplay with the Glyoxalase System. Life, 14(2), 263. https://doi.org/10.3390/life14020263