

Effects of Land Use Type Transformation on the Structure and Diversity of Soil Bacterial Communities


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Sample Collection
2.3. Analysis of Soil Physicochemical Properties
2.4. Sample DNA Extraction
2.5. 16RsRNA Sequence Amplification and Library Construction
2.6. Sequencing Data Processing and Analysis
3. Results
3.1. Physicochemical Properties of the Soil
3.2. Microbial α-Diversity Analysis
3.3. Structural Composition of Microbial Communities
3.4. Correlation between Soil Physical and Chemical Properties and the Relative Abundance of Microbial Communities
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Land Use Types | Sequences | Average Length | Coverage | OTU |
|---|---|---|---|---|
| NF | 110,686 | 263.3 | 0.9775 | 5630 |
| AF | 109,092 | 263.2 | 0.9714 | 5102 |
| FL | 114,581 | 263.6 | 0.9776 | 5821 |
| Phylum | NF | AF | FL |
|---|---|---|---|
| Myxococcota | 0.034 ± 0.003 a | 0.022 ± 0.005 b | 0.031 ± 0.006 ab |
| Acidobacteriota | 0.175 ± 0.171 b | 0.312 ± 0.06 a | 0.146 ± 0.014 b |
| Actinobacteriota | 0.14 ± 0.015 ab | 0.113 ± 0.023 b | 0.187 ± 0.008 a |
| Chloroflexi | 0.073 ± 0.005 a | 0.038 ± 0.003 b | 0.086 ± 0.007 a |
| Bacteroidota | 0.068 ± 0.01 a | 0.061 ± 0.01 a | 0.047 ± 0.002 a |
| Gemmatimonadota | 0.077 ± 0.01 a | 0.024 ± 0.002 b | 0.063 ± 0.002 a |
| Planctomycetota | 0.048 ± 0.002 ab | 0.058 ± 0.004 b | 0.037 ± 0.008 a |
| Proteobacteria | 0.3 ± 0.002 a | 0.315 ± 0.024 a | 0.318 ± 0.019 a |
| Verrucomicrobiota | 0.018 ± 0.004 a | 0.015 ± 0.003 a | 0.016 ± 0.005 a |
| Different Regions | Dominant Soil Bacterial Phylum (Top 4 Abundance) | α Diversity | |||
|---|---|---|---|---|---|
| The results of this study Natural forests | Proteobacteria | Acidobacteriota | Actinobacteriota | Chloroflexi | Agricultural land had the highest Chao1 index, while natural forests had the lowest values. In addition, the Shannon diversity index was highest in natural forests and lowest in agricultural lands. |
| Artificial forests | Proteobacteria | Acidobacteriota | Actinobacteriota | Chloroflexi | |
| Farmland | Proteobacteria | Acidobacteriota | Actinobacteriota | Chloroflexi | |
| Sichuan [23] Sandy wetland | Proteobacteria | Actinobacteriota | Acidobacteriota | Chloroflexi | Chao1 index showed muddy wetlands > sandy wetlands > natural forest land > farmland |
| Farmland | Proteobacteria | Actinobacteriota | Acidobacteriota | Chloroflexi | |
| mud wetland | Proteobacteria | Actinobacteriota | Acidobacteriota | Chloroflexi | |
| natural forest land | Proteobacteria | Actinobacteriota | Acidobacteriota | Chloroflexi | |
| Guizhou [21] bare soil | Proteobacteria | Acidobacteriota | Chloroflexi | Actinobacteriota | The Chao1 and α diversity of soil bacteria in grassland are both at high levels and significantly higher than those in other land use types. and significantly higher than other land use types. The abundance and alpha diversity of soil bacteria in grassland are both at high levels and significantly higher than those in other land use types. and significantly higher than other land use types. |
| Farmland | Proteobacteria | Acidobacteriota | Chloroflexi | Actinobacteriota | |
| Grassland | Proteobacteria | Acidobacteriota | Chloroflexi | Actinobacteriota | |
| Shurb | Proteobacteria | Acidobacteriota | Chloroflexi | Actinobacteriota | |
| GuangXi [20] Farmland | Actinobacteriota | Proteobacteria | Acidobacteriota | Chloroflexi | No significant differences in the richness and diversity indices were found among the three plots. Chao, Simpson, and Shannon diversity indices were the highest in farmland, followed by grassland and plantations. |
| Grass land | Actinobacteriota | Proteobacteria | Acidobacteriota | Chloroflexi | |
| Artificial forest | Actinobacteriota | Proteobacteria | Acidobacteriota | Chloroflexi | |
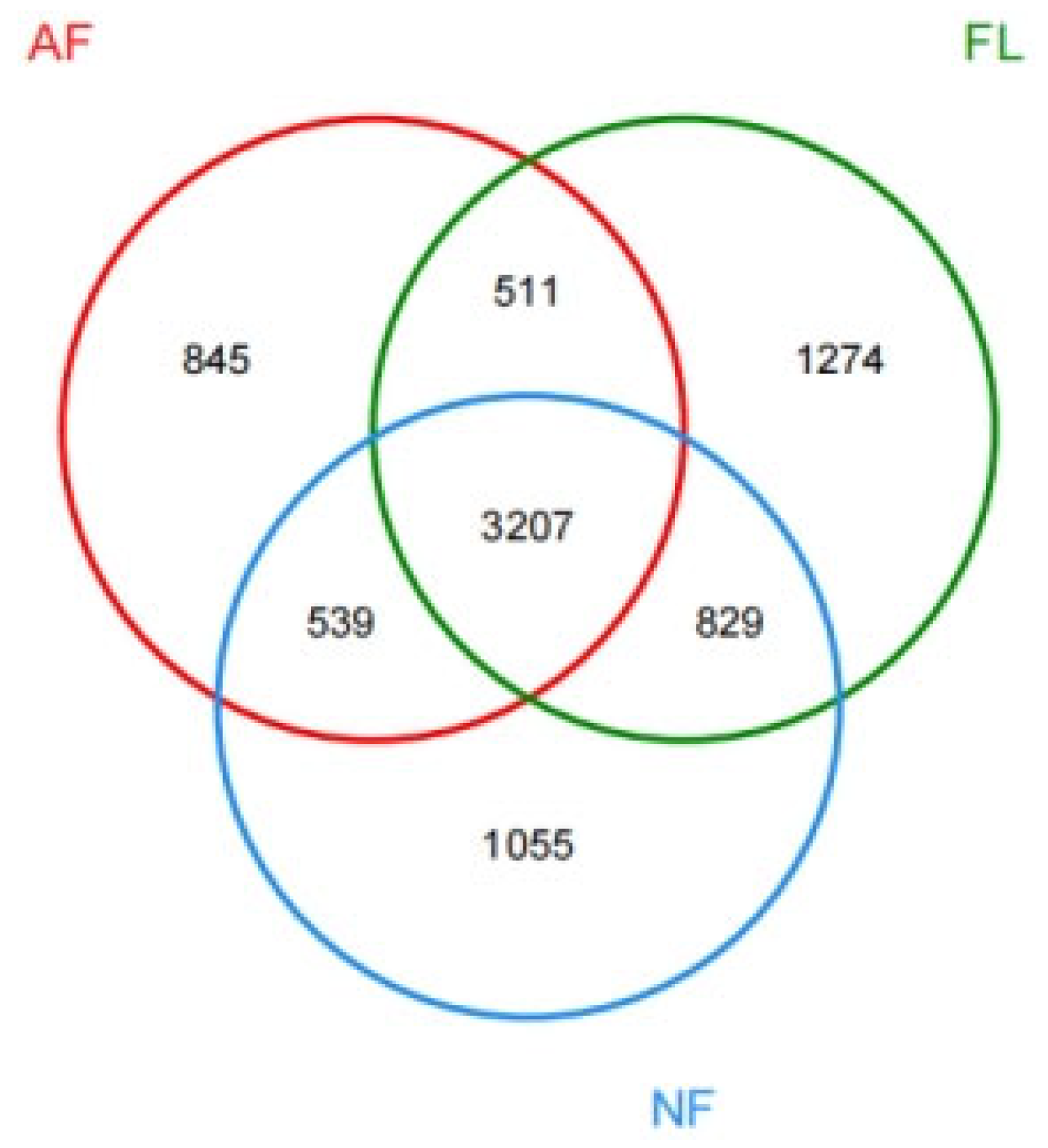
References
- Sun, R.; Sun, B.H.; Gao, M.X.; Yang, X.Y.; Zhang, S.L. Changes of soil microbial characteristics under long-term different land use patterns on an anthropogenic loess soil. J. Plant Nutr. Fertil. 2015, 21, 655–663. [Google Scholar]
- Mganga, K.Z.; Musimba, N.K.R.; Nyariki, D.M. Combining Sustainable Land Management Technologies to Combat Land Degradation and Improve Rural Livelihoods in Semi-arid Lands in Kenya. Environ. Manag. 2015, 56, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, J.L. Bacterial Community Analysis of Panax quinquefolium Rhizosphere Soil by High-throughput Sequencing Technology. J. Chin. Med. Mater. 2019, 42, 7–12. [Google Scholar]
- Sui, X.; Zhang, R.T.; Yang, L.B.; Xu, N.; Chai, C.R.; Wang, J.F.; Fu, X.L.; Zhong, H.X.; Xing, J.H.; Zhang, Y.; et al. Effect of simulation nitrogen depositions on bacterial diversity of Deyeuxia angustifolia in wetland of Sanjiang Plain. Pratacult. Sci. 2016, 33, 589–598. [Google Scholar]
- Yang, J.; Zhou, G.Y.; Tian, Y.Y.; Liu, Q.L.; Liu, C.F.; Yang, Q.; Zhou, C.J. Differential analysis of soil bacteria diversity in different mixed forests of Dalbergia odorifera. J. Ecol. 2015, 35, 8117–8127. [Google Scholar]
- Paletto, A.; Meo, I.D.; Cantiani, P.; Guerrini, S.; Lagomarsino, A. Social perception of forest management: The case of the peri-urban forest of Monte Morello. Forest@ J. Silvicult. For. Ecol. 2018, 15, 29–39. [Google Scholar] [CrossRef]
- He, Z.; Yuan, C.L.; Chen, P.R.; Rong, Z.Q.; Peng, T.; Taimoor, H.F.; Wang, G.J.; Yan, W.D.; Wang, J. Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin. Forests 2023, 14, 1004. [Google Scholar] [CrossRef]
- Yan, B.; Li, J.C.; Xiao, N.W.; Yue, Q.; Fu, G.; Liu, G.H.; Qiao, M.P. Urban-development-induced Changes in the Diversity and Composition of the Soil Bacterial Community in Beijing. Sci. Rep. 2016, 6, 38811. [Google Scholar] [CrossRef]
- Zhou, J.Z.; He, Z.; Yang, Y.F.; Deng, Y.; Tringe, S.; Cohen, L.A. High-Throughput Metagenomic Technologies for Complex Microbial Community Analysis: Open and Closed Formats. mBio 2015, 6, e02288-14. [Google Scholar] [CrossRef]
- Li, J.B.; Pokharel, P.; Liu, G.M.; Chen, J.L. Reclamation of desert land to different land-use types changes soil bacterial community composition in a desert-oasis ecotone. Land Degrad. Dev. 2020, 32, 1389–1399. [Google Scholar] [CrossRef]
- Wu, Y.N.; Gao, W.F.; Zou, Y.; Dong, H.Y.; Yu, F.; Wang, H.; Cheng, Z. Effects of Land Use Conversion on the Soil Microbial Community Composition and Functionality in the Urban Wetlands of North-Eastern China. Forests 2022, 13, 1148. [Google Scholar] [CrossRef]
- He, X.Y.; Su, Y.R.; Liang, Y.M.; Chen, X.B.; Zhu, H.H.; Wang, K.L. Land reclamation and short-term cultivation change soil microbial communities and bacterial metabolic profiles. J. Sci. Food Agric. 2012, 92, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Yang, J.H.; Xie, X.F.; Chen, X.J.; Pu, L.J.; Zhang, X.W. Soil microbial succession with soil development since costal reclamation. Catena 2020, 187, 104393. [Google Scholar] [CrossRef]
- Wang, D.W.; Wang, J.; Fu, G. Dynamic Assessment and Analysis of Grain Production Balance: A Study on Strategies for the Sustainable Development of Grain Security in the Western Region of Heilongjiang Province, China. Hydra Sci. Tech. 2007, 6, 9–10. [Google Scholar]
- Cheng, Z.Q.; Yang, L.B.; Zhang, T.; Wang, W.H.; Yin, W.P.; Li, G.F.; Song, F.Q. Soil Microbial Functional Diversity Responses to Different Revegetation Types in Heilongjiang Zhongyangzhan Black-Billed Capercaillie Nature Reserve. Res. Environ. Sci. 2021, 34, 1177–1186. [Google Scholar]
- Jia, P.L.; An, S.S.; Li, C.C.; Zeng, Q.C.; Wang, B.R.; Bai, X.J. Dynamics of Soil Nutrients and Their Ecological Stoichiometry Characteristics Under Different Longitudes in the East-West Forest Belt of Loess Plateau. J. Soil Water Conserv. 2020, 34, 315–321. [Google Scholar]
- Yu, J.Y.; Huang, X.H.; Shuai, Z.B.; Xie, L.; Liu, Y.H.; Wang, C.R.; Chai, D.; Liu, D.; Zhou, X.Y.; Chen, Q. The community structure of bacteria and fungi in soils with root rot diseased garlic plants in Pengzhou, Sichuan Province. Chin. J. Appl. Environ. Biol. 2020, 26, 928–935. [Google Scholar]
- Chen, X.R.; Nan, B.Z. Bacterial Diversity and Its Role in Agricultural Ecosystems. Pratacult. Sci. 2002, 9, 34–38. [Google Scholar]
- Zhao, F.; Zhao, M.Z.; Wang, J.; Guan, L.; Pang, F.H. Studying the Microbial Community Structure and Diversity in the Rhizosphere of Strawberries Using High-Throughput Sequencing. Soils 2019, 51, 51–60. [Google Scholar]
- Zou, X.X.; Yao, K.; Zeng, F.P.; Zhang, C.; Yao, Z.X.; Zhang, H. Diversity and Assembly of Bacteria Community in Lime Soil under Different Karst Land-Use Types. Forests 2023, 14, 672. [Google Scholar] [CrossRef]
- Cai, G.X.; Xia, F.; Li, D.; Bao, Q.; Chen, B.; Zhao, M. A Simulation Study on Characteristics of Soil Microbial Community Structure under Different Land Uses. Bull. Miner. Petrol. Geochem. 2022, 41, 1023–1032. [Google Scholar]
- Qin, H.; Li, C.X.; Ren, Q.S. Effects of different land use patterns on soil bacterial and fungal biodiversity in the hydro-fluctuation zone of the Three Gorges reservoir region. Acta Ecol. Sin. 2017, 37, 3494–3504. [Google Scholar]
- Liu, K.H.; Xue, Y.Q.; Zhu, L.P.; Xu, F.; Zhu, Z.H.; Zhang, T.; Zhang, F.B. Effect of Different Land Use Types on the Diversity of Soil Bacterial Community in the Coastal Zone of Jialing River. Acta Ecol. Sin. 2022, 43, 1620–1629. [Google Scholar]
- Foulon, J.; Zappelini, C.; Durand, C.; Valot, B.; Girardclos, O.; Blaudez, D.; Chalot, M. Environmental metabarcoding reveals contrasting microbial communities at two poplar phytomanagement sites. Sci. Total Environ. 2016, 571, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, S.K.; Gao, Q.Z.; Liu, S.L.; Zhou, H.K.; Ganjurjav, H.; Wang, X.X. Climate change and human activities altered the diversity and composition of soil microbial community in alpine grasslands of the Qinghai-Tibetan Plateau. Sci. Total Environ. 2016, 562, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, G.B.; Sha, X.; Wang, G.L. Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Sci. Total Environ. 2016, 97, 40–49. [Google Scholar] [CrossRef]
- Ding, H.; Fang, Y.M.; Yang, Q.; Chen, X.; Yuan, F.Y.; Xu, H.; He, L.H.; Yan, J.; Chen, T.T.; Yu, C.J.; et al. Community characteristics of a mid-subtropical evergreen broad-leaved forest plot in the Wuyi Mountains, Fujian Province, Southeastern China. Biodivers. Sci. 2015, 4, 479–492. [Google Scholar] [CrossRef]
- Liu, C.X.; Dong, Y.H.; Jiao, R.Z. Research Progress in Acidobacteria Diversity in Forest Soil. World For. Res. 2016, 29, 17–22. [Google Scholar]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an Ecological Classification of Soil Bacteria. Bull. Ecol. Soc. Am. 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Shen, L.; Li, Z.H.; Zeng, W.M.; Yu, R.L.; Wu, X.L.; Li, J.K.; Wang, L.K. Microbial Communities in Soils of Qingshuitang Industrial District in Zhuzhou. Acta Ecol. Sin. 2018, 39, 5151–5162. [Google Scholar]
- Huang, S.; Pan, X.H.; Huang, Q.R.; Yu, X.C.; Li, C.X.; Zhang, W.J. Effect of Long-term Fertilization in Different Patterns on the Productivity and Soil Structure of Red Hilly Upland in Southern China. Acta Agric. Univ. Jiangxiensis 2012, 34, 403–408. [Google Scholar]
- Costello, E.K.; Schmidt, S.K. Microbial diversity in alpine tundra wet meadow soil: Novel Chloroflexi from a cold, water-saturated environment. Environ. Microbiol. 2006, 8, 1471–1486. [Google Scholar] [CrossRef]
- Cohen, J.K.; Zolti, A.; Harpaz, L.S.; Argaman, E.; Rabinovich, R.; Green, S.J.; Minz, D. Effects of tillage practices on soil microbiome and agricultural parameters. Sci. Total Environ. 2020, 705, 135791. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Ren, Y.R.; Liao, B.A.; Dong, Q.; Liu, M.S. Composition and structural characteristics of soil microbial communities in Yancheng typical coastal wetlands. Acta Ecol. Sin. 2023, 43, 2336–2347. [Google Scholar]
- Jiao, S.; Chen, W.M.; Wang, Q.; Du, N.N.; Li, Q.P.; Wei, G.H. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Curr. Stem Cell Res. Ther. 2018, 6, 146. [Google Scholar] [CrossRef]
- Zeng, J.; Shen, J.P.; Wang, J.T.; Hu, H.W.; Zhang, C.J.; Bai, R.; Zhang, L.W.; He, J.Z. Impacts of Projected Climate Warming and Wetting on Soil Microbial Communities in Alpine Grassland Ecosystems of the Tibetan Plateau. Microb. Ecol. 2018, 75, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Z.; Cai, Z.C. Effect of Agricultural Cultivation on Soil Organic Carbon in China. J. Soil Water Conserv. 2007, 21, 118–121+134. [Google Scholar]
- Wang, G.Z.; Liu, Y.G.; Cui, M.; Zhou, Z.Y.; Zhang, Q.; Li, Y.J.; Ha, W.X.; Pang, D.B.; Luo, J.F.; Zhou, J.X. Effects of secondary succession on soil fungal and bacterial compositions and diversities in a karst area. Plant Soil 2021, 475, 1–12. [Google Scholar] [CrossRef]
- George, P.B.L.; Lallias, D.; Creer, S.; Seaton, F.M.; Kenny, J.G.; Eccles, R.M.; Griffiths, R.I.; Lebron, I.; Emmett, B.A.; Robinson, D.A.; et al. Divergent national-scale trends of microbial and animal biodiversity revealed across diverse temperate soil ecosystems. Nat. Commun. 2019, 10, 1107. [Google Scholar] [CrossRef] [PubMed]
- Lammel, D.R.; Nüsslein, K.; Veresoglou, C.E.P.C.S.D.; Rillig, M.C. Soil biota shift with land use change from pristine rainforest and Savannah (Cerrado) to agriculture in southern Amazonia. Mol. Ecol. 2021, 30, 4899–4912. [Google Scholar] [CrossRef]
- Santillan, E.; Seshan, H.; Constancias, F.; Moses, D.I.D.; Wuertz, S. Frequency of disturbance alters diversity, function, and underlying assembly mechanisms of complex bacterial communities. npj Biofilms Microbiomes 2019, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.H.; Li, R.; Zhang, L.L. Soil nutrient variability mediates the effects of erosion on soil microbial communities: Results from a modified topsoil removal method in an agricultural field in Yunnan plateau, China. Environ. Sci. Pollut. Res. Int. 2021, 29, 3659–3671. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Marschner, P.; Cao, W.H.; Zuo, C.Q.; Qin, W. Influence of salinity and water content on soil microorganisms. Int. Soil Water Conserv. Res. 2015, 3, 316–323. [Google Scholar] [CrossRef]
- Liu, J.K.; Engel, A.B.; Wang, Y.; Zhang, G.F.; Zhang, Z.M.; Zhang, M.X. Multi-scale analysis of hydrological connectivity and plant response in the Yellow River Delta. Sci. Total Environ. 2020, 702, 134889. [Google Scholar] [CrossRef]
- Daniel, L.F.; Eugen, U.; Mihaela, B.A.; Feodor, F.; Cristina, M.E. Diversity of soil bacteria as Indicator of Soil Pollution in Moldavia Region, Romania. Environ. Eng. Manag. J. 2017, 16, 879–889. [Google Scholar]
- Steenwerth, K.L. Links between Soil Microbial Communities and Transformations of Soil Carbon and Nitrogen along a Gradient in Land-Use History and Soil Disturbance. Ph.D. Thesis, University of California, Davis, CA, USA, 2003. [Google Scholar]
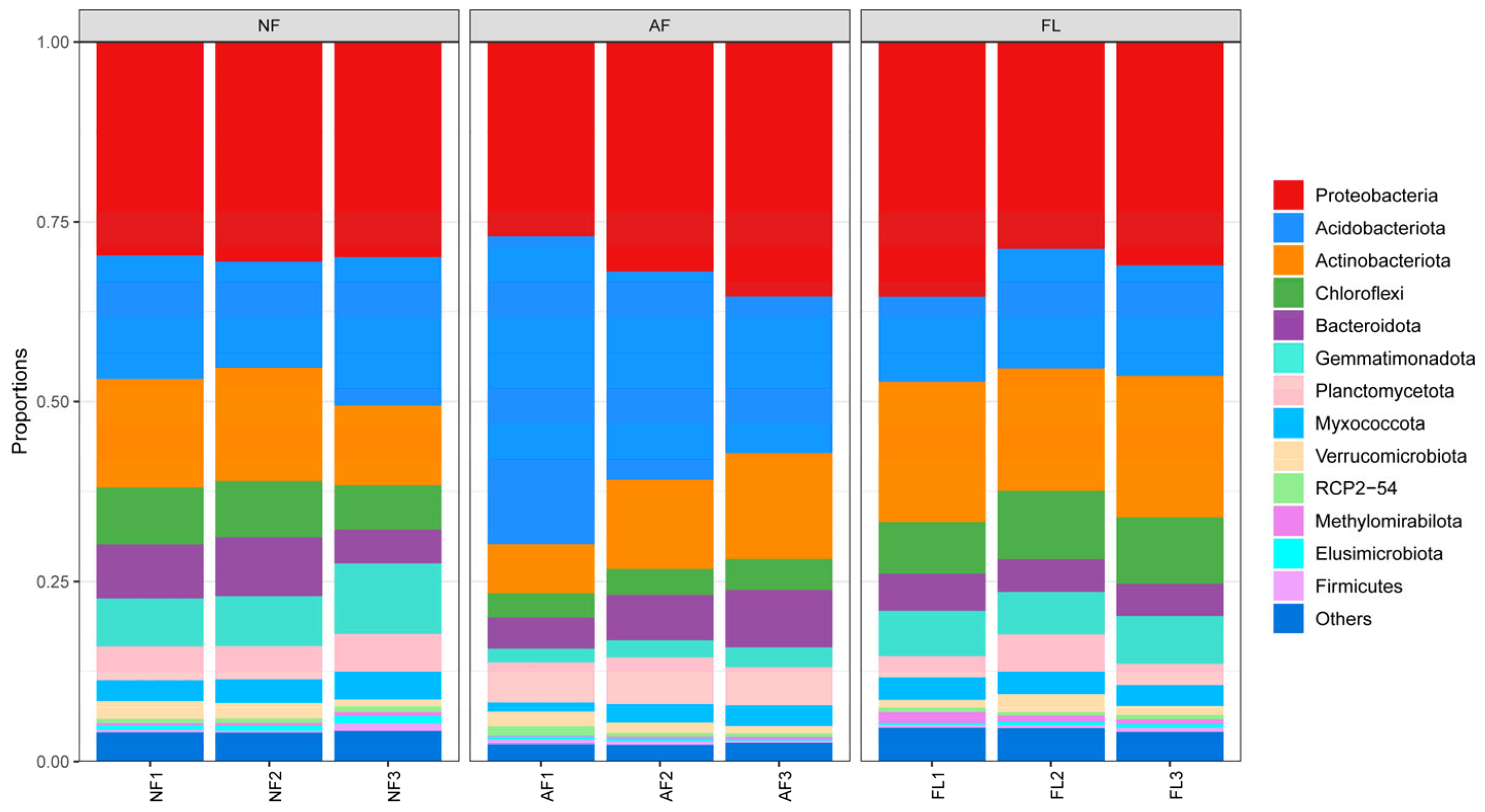
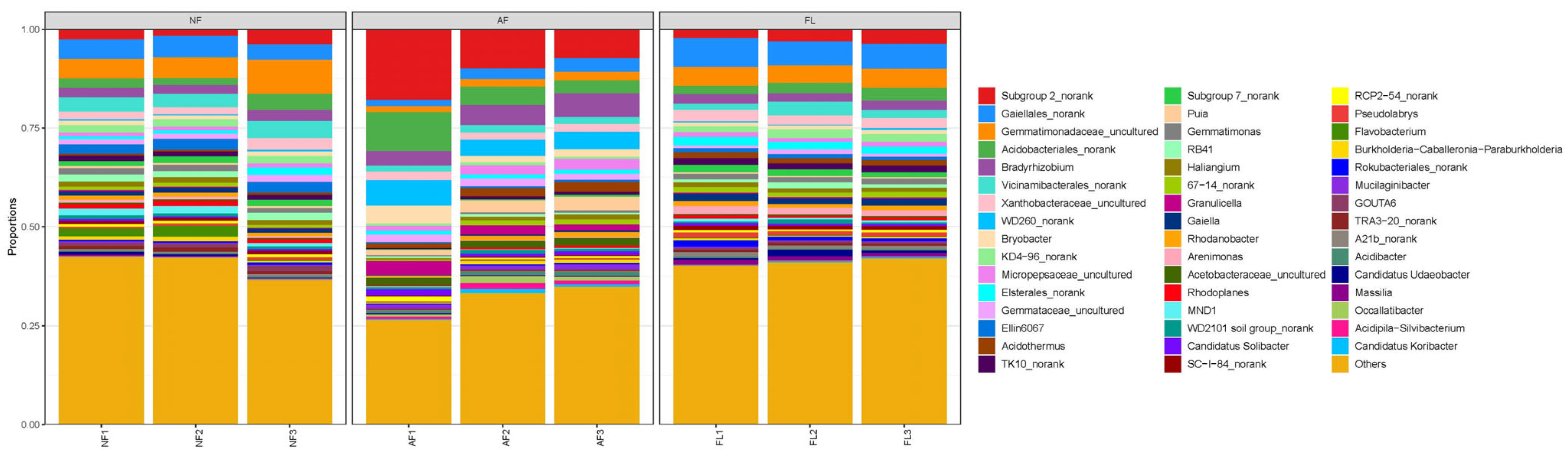
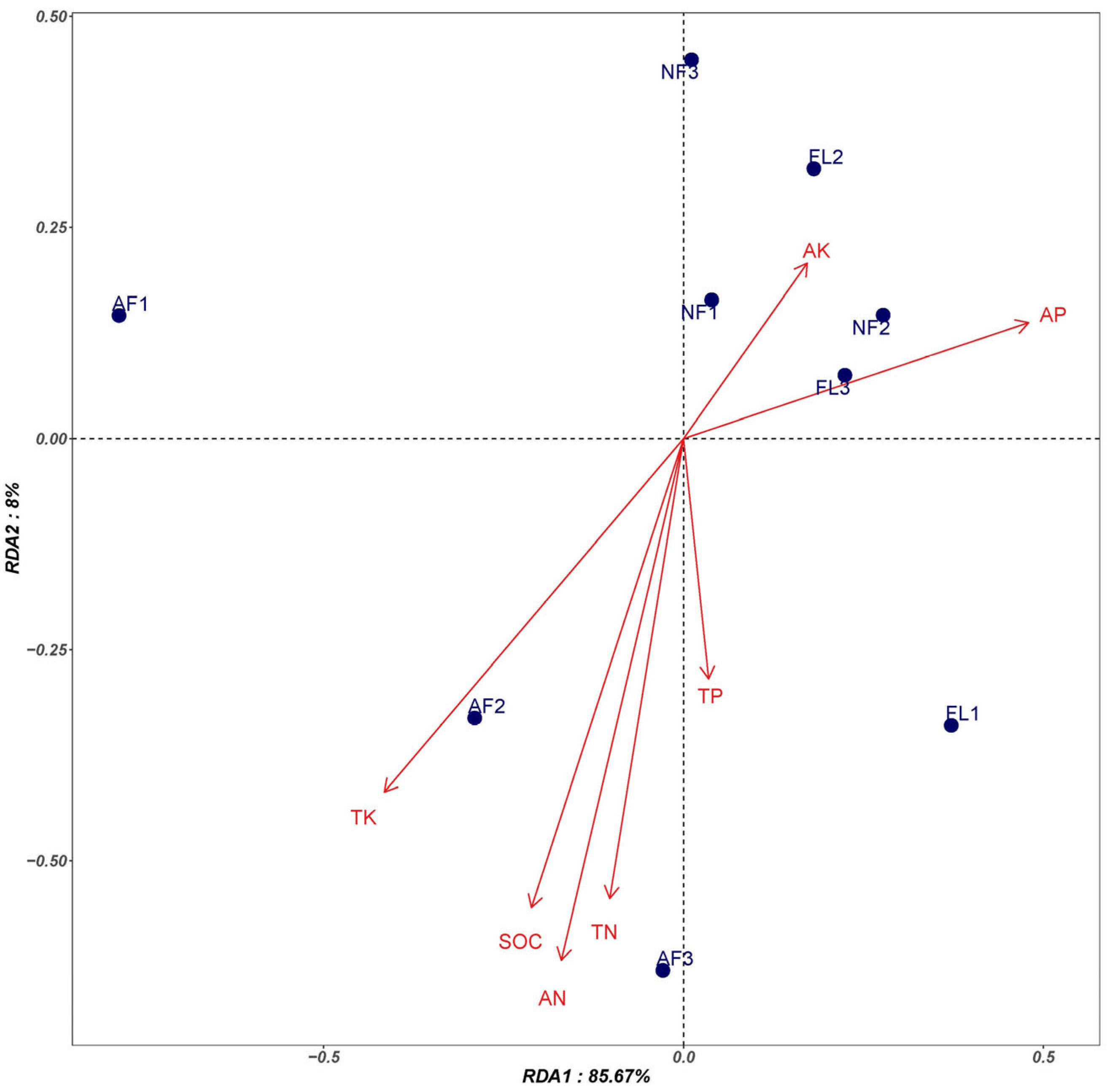
| Soil Sample | TN (g/kg) | AN (mg/kg) | TP (g/kg) | AP (mg/kg) | TK (g/kg) | AK (mg/kg) | SOC (g/kg) |
|---|---|---|---|---|---|---|---|
| NF | 1.672 ± 0.159 b | 85.167 ± 5.09 b | 0.264 ± 0.02 a | 5.32 ± 0.67 b | 6.25 ± 0.28 b | 61.80 ± 4.32 a | 71.47 ± 7.11 b |
| AF | 4.166 ± 0.948 a | 156.333 ± 21.61 a | 0.479 ± 0.06 b | 5.24 ± 0.85 b | 9.76 ± 0.62 a | 64.08 ± 20.46 a | 64.23 ± 4.65 a |
| FL | 3.155 ± 0.464 ab | 135.333 ± 9.33 a | 0.586 ± 0.32 b | 30.76 ± 3.72 a | 9.31 ± 0.334 a | 86.42 ± 18.04 a | 43.64 ± 3.77 a |
| Land Use Type | Chao1 | Shannon | Simpson | Coverage |
|---|---|---|---|---|
| NF | 4343.4 ± 14.939 a | 10.2 ± 0.108 a | 0.027 ± 0.001 b | 0.974 ± 0.005 a |
| AF | 3775.5 ± 159.176 b | 10.41 ± 0.198 a | 0.557 ± 0.011 a | 0.974 ± 0.002 a |
| FL | 4475.6 ± 116.811 a | 9.6 ± 0.3555 b | 0.193 ± 0.003 b | 0.973 ± 0.003 a |
| Phylum | Physical and Chemical Properties of the Soil | ||||||
|---|---|---|---|---|---|---|---|
| AK | AN | TN | TP | TK | AP | SOC | |
| Myxococcota | 0.2333 | −0.569 | −0.4833 | −0.2333 | −0.6667 * | 0.35 | −0.5167 |
| Acidobacteriota | −0.2 | 0.2343 | 0.2333 | −0.05 | 0.3833 | −0.5333 | 0.4 |
| Actinobacteriota | 0.5 | 0.159 | 0.1333 | 0.45 | −0.1333 | 0.6833 * | −0.1 |
| Chloroflexi | 0.4667 | −0.1255 | −0.0833 | 0.2333 | −0.4 | 0.5167 | −0.1 |
| Bacteroidota | −0.3667 | −0.0084 | −0.15 | −0.5333 | −0.5833 | −0.2833 | −0.1353 |
| Gemmatinnonadota | 0.1333 | −0.7029 * | −0.6833 * | −0.333 | −0.8333 ** | 0.1167 | −0.75 * |
| Planctomycetota | 0.0833 | 0.3264 | 0.3533 | −0.0333 | 0.35 | −0.3833 | 0.5 |
| Proteobacteria | −0.05 | 0.4268 | 0.3333 | 0.25 | −0.0167 | 0.2667 | 0.3667 |
| Verrucomicrobiota | 0.1833 | 0.0418 | 0.0667 | −0.0167 | 0.2167 | 0.0167 | −0.1167 |
| Chao1 | 0.35 | −0.0335 | −0.05 | 0.2833 | −0.3833 | 0.4833 | −0.3167 |
| Shannon | 0.4333 | −0.1506 | −0.1333 | 0.25 | −0.4167 | 0.5 | −0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, H.; Sui, X.; Liu, Y.; Liu, X.; Chang, Q.; Xu, R.; Li, M.; Mu, L. Effects of Land Use Type Transformation on the Structure and Diversity of Soil Bacterial Communities. Life 2024, 14, 252. https://doi.org/10.3390/life14020252
Hua H, Sui X, Liu Y, Liu X, Chang Q, Xu R, Li M, Mu L. Effects of Land Use Type Transformation on the Structure and Diversity of Soil Bacterial Communities. Life. 2024; 14(2):252. https://doi.org/10.3390/life14020252
Chicago/Turabian StyleHua, Henian, Xin Sui, Yanan Liu, Xu Liu, Qiuyang Chang, Ruiting Xu, Mengsha Li, and Liqiang Mu. 2024. "Effects of Land Use Type Transformation on the Structure and Diversity of Soil Bacterial Communities" Life 14, no. 2: 252. https://doi.org/10.3390/life14020252
APA StyleHua, H., Sui, X., Liu, Y., Liu, X., Chang, Q., Xu, R., Li, M., & Mu, L. (2024). Effects of Land Use Type Transformation on the Structure and Diversity of Soil Bacterial Communities. Life, 14(2), 252. https://doi.org/10.3390/life14020252