

Astaxanthin Protects Against H2O2- and Doxorubicin-Induced Cardiotoxicity in H9c2 Rat Myocardial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Viability Analysis
2.3. Confocal Microscopy
2.4. Analysis of Cytosolic Ca2+ Content
2.5. Western Blot Assay
2.6. Statistical Analysis
3. Results
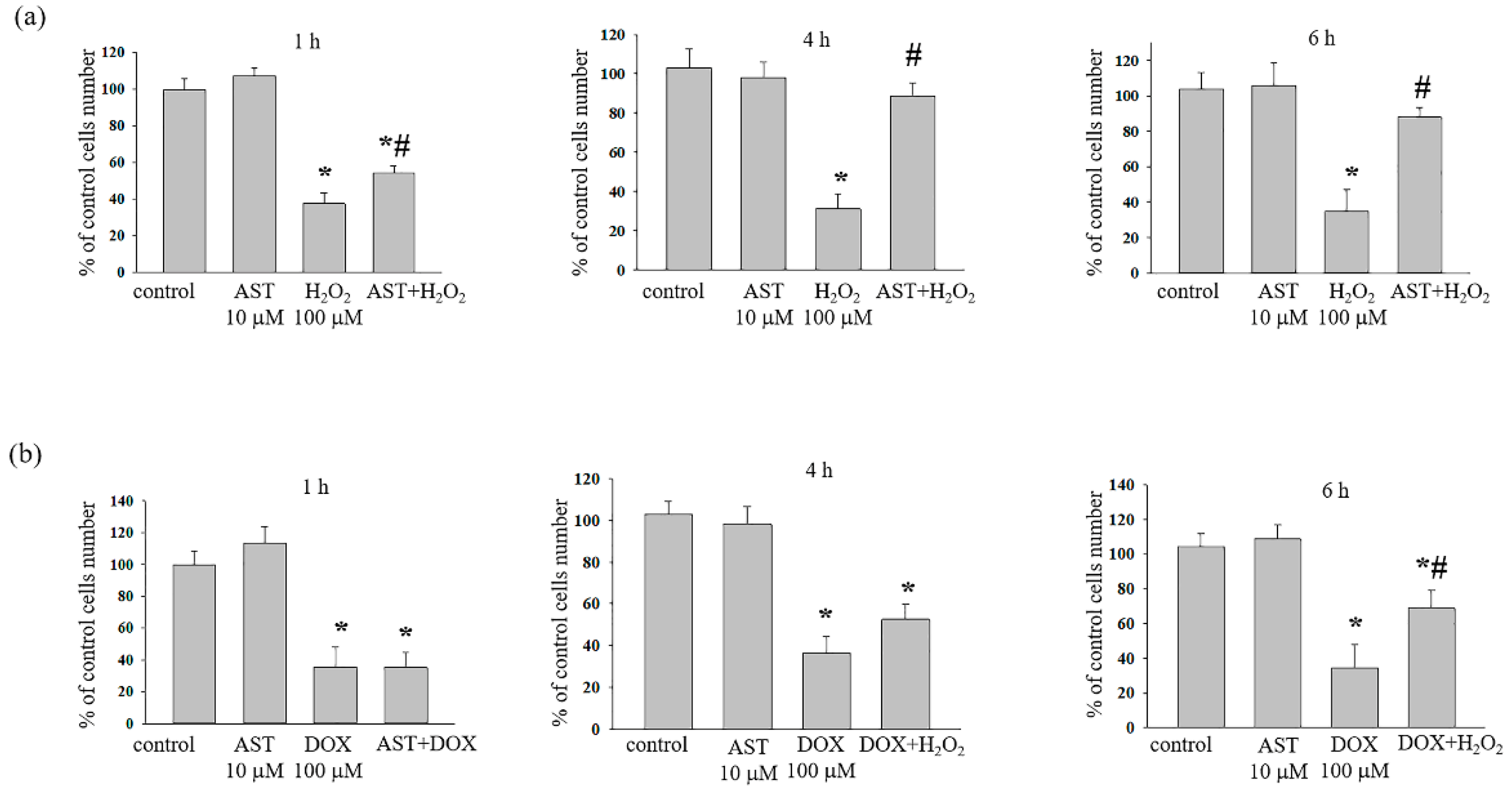
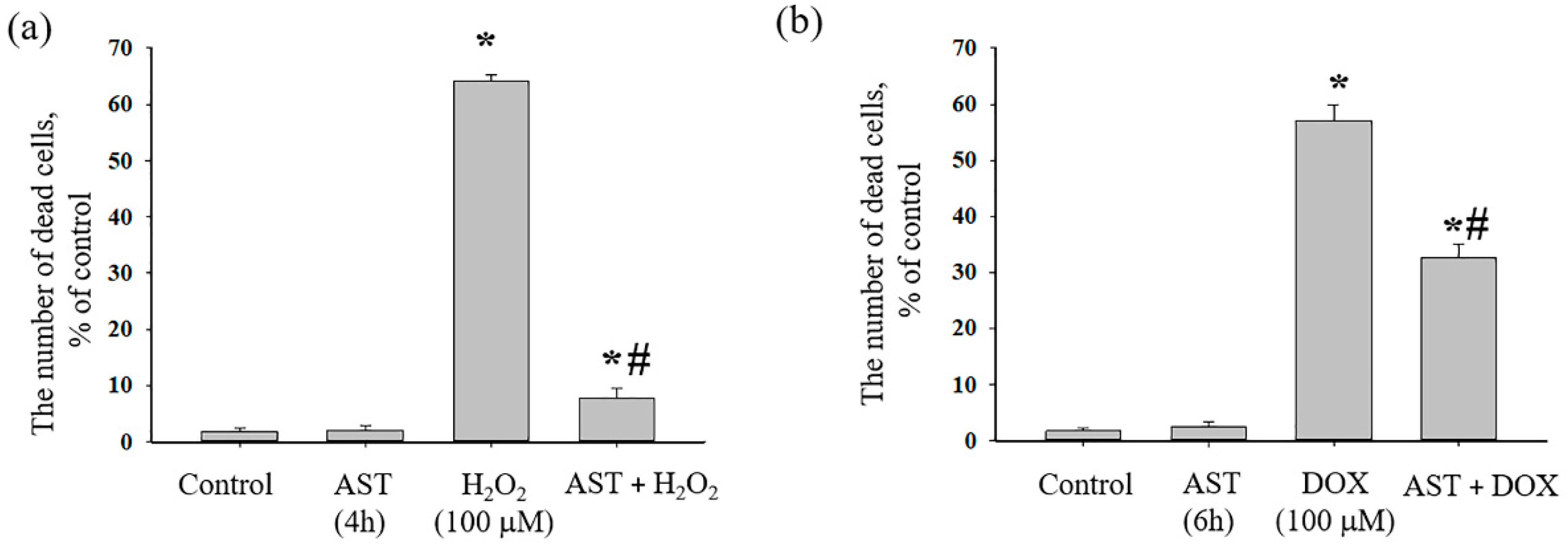
3.1. The Cytotoxic Effect of AST, H2O2, and DOX on the Survival of H9c2 Cardiomyocytes
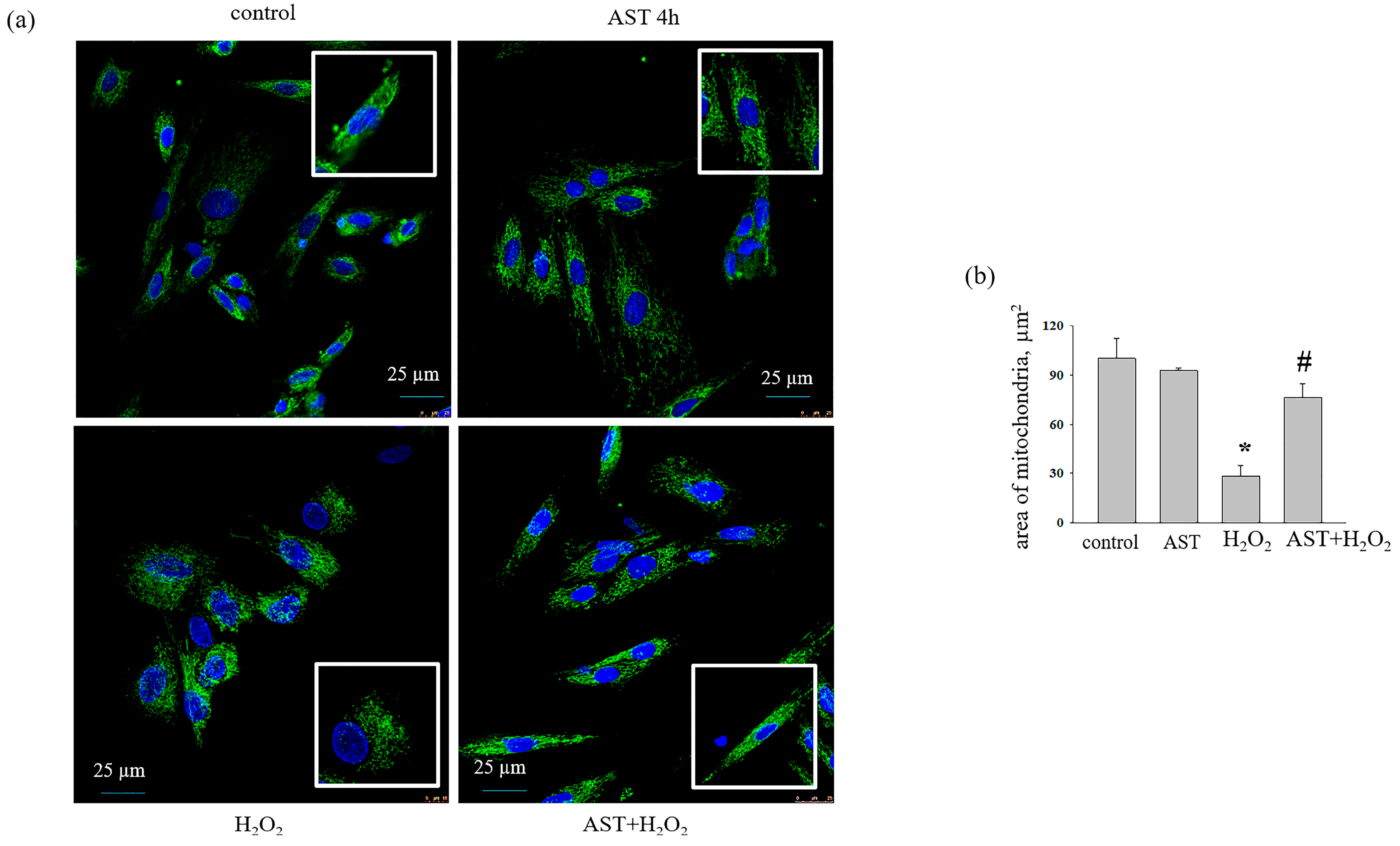
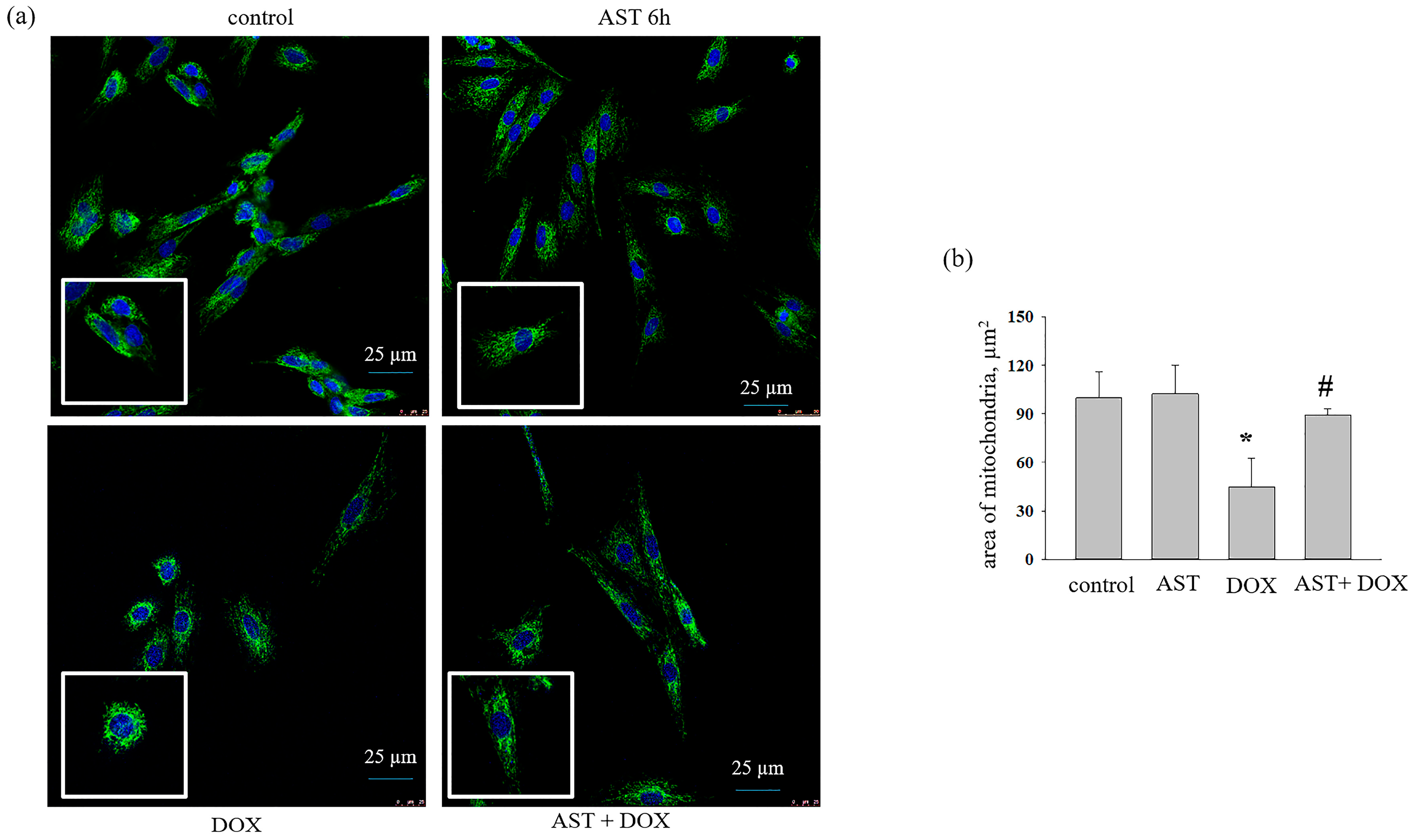
3.2. The Effect of AST, H2O2, and DOX on the Change in Mitochondrial Mass in H9c2 Cardiomyocytes
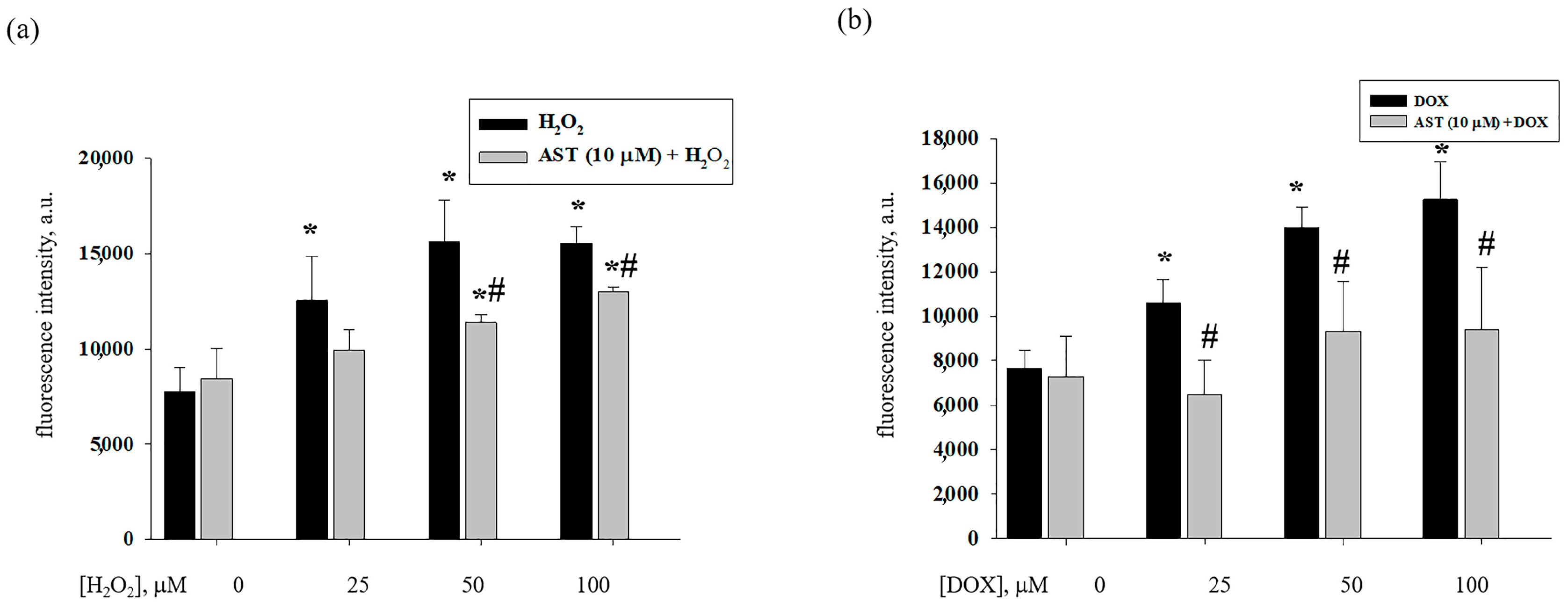
3.3. Effect of AST, H2O2, and DOX on the Content of Cytosolic Ca2+ in H9c2 Cardiomyocytes
3.4. Effect of AST, H2O2, and DOX on the Level of VDAC1 and CHOP in H9c2 Cardiomyocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, S.; Koo, S. Efficient syntheses of the keto-carotenoids canthaxanthin, astaxanthin, and astacene. J. Org. Chem. 2005, 70, 3328–3331. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Bassi, A. Carotenoids from microalgae: A review of recent developments. Biotechnol. Adv. 2016, 34, 1396–1412. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Abdelzaher, L.A.; Imaizumi, T.; Suzuki, T.; Tomita, K.; Takashina, M.; Hattori, Y. Astaxanthin alleviates oxidative stress insults-related derangements in human vascular endothelial cells exposed to glucose fluctuations. Life Sci. 2016, 150, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory effect of astaxanthin on oxidative stress-induced mitochondrial dysfunction-a mini-review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef]
- Krestinin, R.; Baburina, Y.; Odinokova, I.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Krestinina, O. Isoproterenol-induced permeability transition pore-related dysfunction of heart mitochondria is attenuated by astaxanthin. Biomedicines 2020, 8, 437. [Google Scholar] [CrossRef]
- Krestinina, O.; Baburina, Y.; Krestinin, R.; Odinokova, I.; Fadeeva, I.; Sotnikova, L. Astaxanthin prevents mitochondrial impairment induced by isoproterenol in isolated rat heart mitochondria. Antioxidants 2020, 9, 262. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De, S.; Meir, A. The mitochondrial voltage-dependent anion channel 1, Ca2+ transport, apoptosis, and their regulation. Front. Oncol. 2017, 7, 60. [Google Scholar] [CrossRef]
- Schwertz, H.; Carter, J.M.; Abdudureheman, M.; Russ, M.; Buerke, U.; Schlitt, A.; Muller-Werdan, U.; Prondzinsky, R.; Werdan, K.; Buerke, M. Myocardial ischemia/reperfusion causes vdac phosphorylation which is reduced by cardioprotection with a p38 map kinase inhibitor. Proteomics 2007, 7, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, H.; Chen, G.; Feng, Y.; Zou, J.; Liu, M.; Liu, K.; Wang, N.; Zhang, H.; Wang, K.; et al. Wdr26/mip2 interacts with vdac1 and regulates vdac1 expression levels in h9c2 cells. Free Radic. Biol. Med. 2018, 117, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xu, Y.; Heisner, J.S.; Sun, J.; Stowe, D.F.; Kwok, W.M.; Camara, A.K.S. Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase ii in mitochondria. Mitochondrion 2019, 46, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Roberts, R.; Marian, A.J. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: Insight into the pathogenesis of phenotypes. J. Am. Coll. Cardiol. 2001, 38, 1175–1180. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Nishitoh, H. Chop is a multifunctional transcription factor in the er stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Jungsuwadee, P. Doxorubicin-induced cardiomyopathy: An update beyond oxidative stress and myocar-dial cell death. Cardiovasc. Regen. Med. 2016, 3, e1127. [Google Scholar]
- Janero, D.R.; Hreniuk, D.; Sharif, H.M. Hydrogen peroxide-induced oxidative stress to the mammalian heart-muscle cell (cardiomyocyte): Lethal peroxidative membrane injury. J. Cell Physiol. 1991, 149, 347–364. [Google Scholar] [CrossRef]
- Chen, Q.M.; Tu, V.C.; Wu, Y.; Bahl, J.J. Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Arch. Biochem. Biophys. 2000, 373, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar] [PubMed]
- Kayikcioglu, M.; Ozkan, H.S.; Yagmur, B. Premature myocardial infarction: A rising threat. Balkan Med. J. 2022, 39, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 2009, 34, 1021–1029. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Ko, J.C.; Chen, J.C.; Wang, T.J.; Zheng, H.Y.; Chen, W.C.; Chang, P.Y.; Lin, Y.W. Astaxanthin down-regulates rad51 expression via inactivation of akt kinase to enhance mitomycin c-induced cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharmacol. 2016, 105, 91–100. [Google Scholar] [CrossRef]
- Faraone, I.; Sinisgalli, C.; Ostuni, A.; Armentano, M.F.; Carmosino, M.; Milella, L.; Russo, D.; Labanca, F.; Khan, H. Astaxanthin anticancer effects are mediated through multiple molecular mechanisms: A systematic review. Pharmacol. Res. 2020, 155, 104689. [Google Scholar] [CrossRef]
- Robin, E.D.; Wong, R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell Physiol. 1988, 136, 507–513. [Google Scholar] [CrossRef]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Fu, X.Y.; Sun, B.L. Astaxanthin attenuates homocysteine-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [PubMed]
- Krestinin, R.; Kobyakova, M.; Baburina, Y.; Sotnikova, L.; Krestinina, O. Astaxanthin reduces h2o2– and doxorubicin-induced cardiotoxocity in h9c2 cardiomyocite cells. Biochem. Mosc. 2024, 89. in press. [Google Scholar]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Lower, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating opa1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D. Vdac, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef]
- Mitra, A.; Basak, T.; Datta, K.; Naskar, S.; Sengupta, S.; Sarkar, S. Role of alpha-crystallin b as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013, 4, e582. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y. Tauroursodeoxycholic acid alleviates h(2)o(2)-induced oxidative stress and apoptosis via suppressing endoplasmic reticulum stress in neonatal rat cardiomyocytes. Dose Response 2018, 16, 1559325818782631. [Google Scholar] [CrossRef]
- Qiu, Y.; Cheng, R.; Liang, C.; Yao, Y.; Zhang, W.; Zhang, J.; Zhang, M.; Li, B.; Xu, C.; Zhang, R. Microrna-20b promotes cardiac hypertrophy by the inhibition of mitofusin 2-mediated inter-organelle Ca2+ cross-talk. Mol. Ther. Nucleic Acids 2020, 19, 1343–1356. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- de Ridder, I.; Kerkhofs, M.; Lemos, F.O.; Loncke, J.; Bultynck, G.; Parys, J.B. The er-mitochondria interface, where Ca2+ and cell death meet. Cell Calcium 2023, 112, 102743. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ero1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krestinin, R.; Kobyakova, M.; Baburina, Y.; Sotnikova, L.; Krestinina, O. Astaxanthin Protects Against H2O2- and Doxorubicin-Induced Cardiotoxicity in H9c2 Rat Myocardial Cells. Life 2024, 14, 1409. https://doi.org/10.3390/life14111409
Krestinin R, Kobyakova M, Baburina Y, Sotnikova L, Krestinina O. Astaxanthin Protects Against H2O2- and Doxorubicin-Induced Cardiotoxicity in H9c2 Rat Myocardial Cells. Life. 2024; 14(11):1409. https://doi.org/10.3390/life14111409
Chicago/Turabian StyleKrestinin, Roman, Margarita Kobyakova, Yulia Baburina, Linda Sotnikova, and Olga Krestinina. 2024. "Astaxanthin Protects Against H2O2- and Doxorubicin-Induced Cardiotoxicity in H9c2 Rat Myocardial Cells" Life 14, no. 11: 1409. https://doi.org/10.3390/life14111409
APA StyleKrestinin, R., Kobyakova, M., Baburina, Y., Sotnikova, L., & Krestinina, O. (2024). Astaxanthin Protects Against H2O2- and Doxorubicin-Induced Cardiotoxicity in H9c2 Rat Myocardial Cells. Life, 14(11), 1409. https://doi.org/10.3390/life14111409