
The Association of Novel Single-Nucleotide Variants in the Collagen Matrix-Encoding Gene PRDM5 with Aortic Aneurysmal Disease

Abstract
1. Introduction
2. Case Descriptions
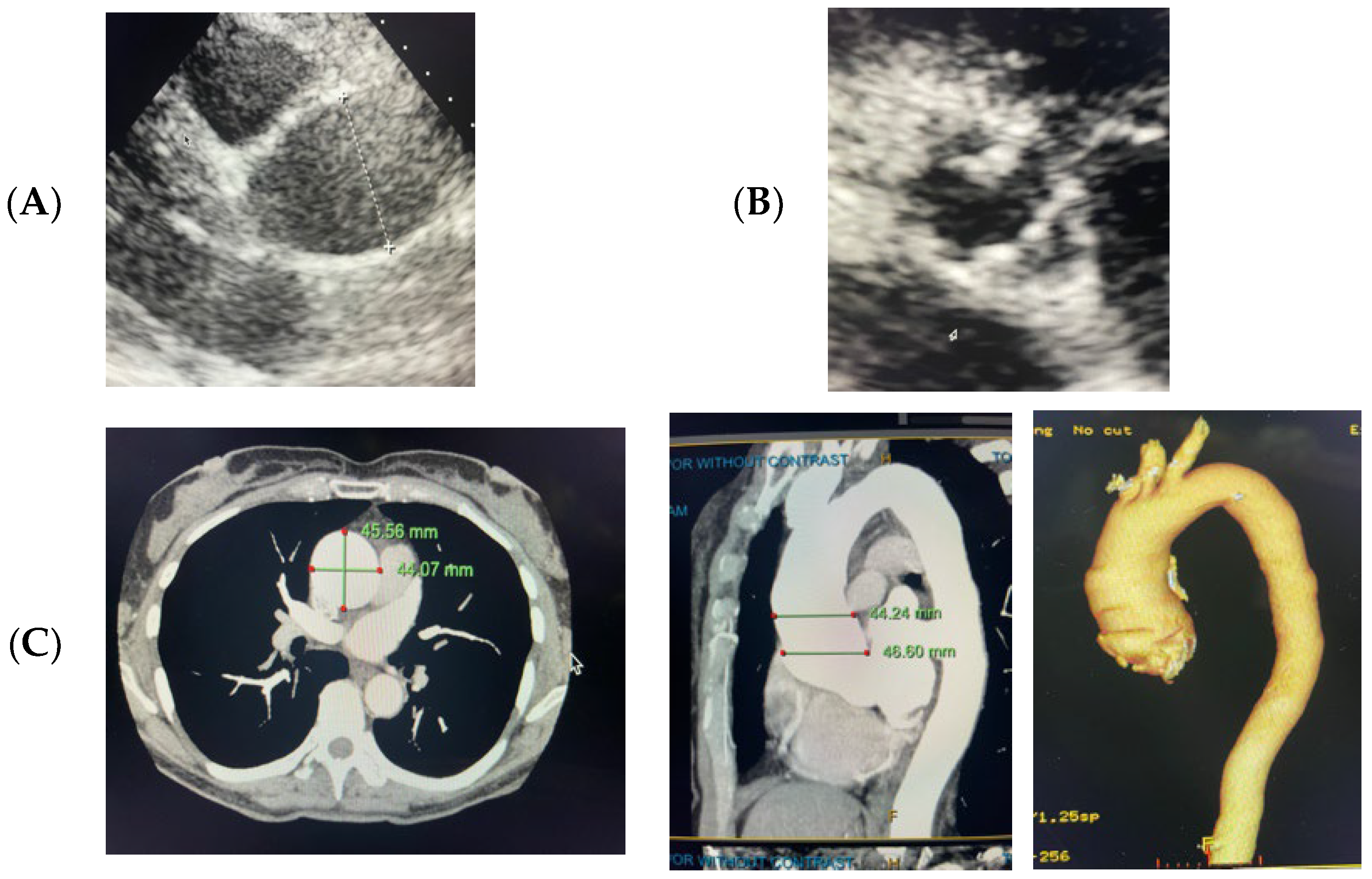
2.1. Case 1
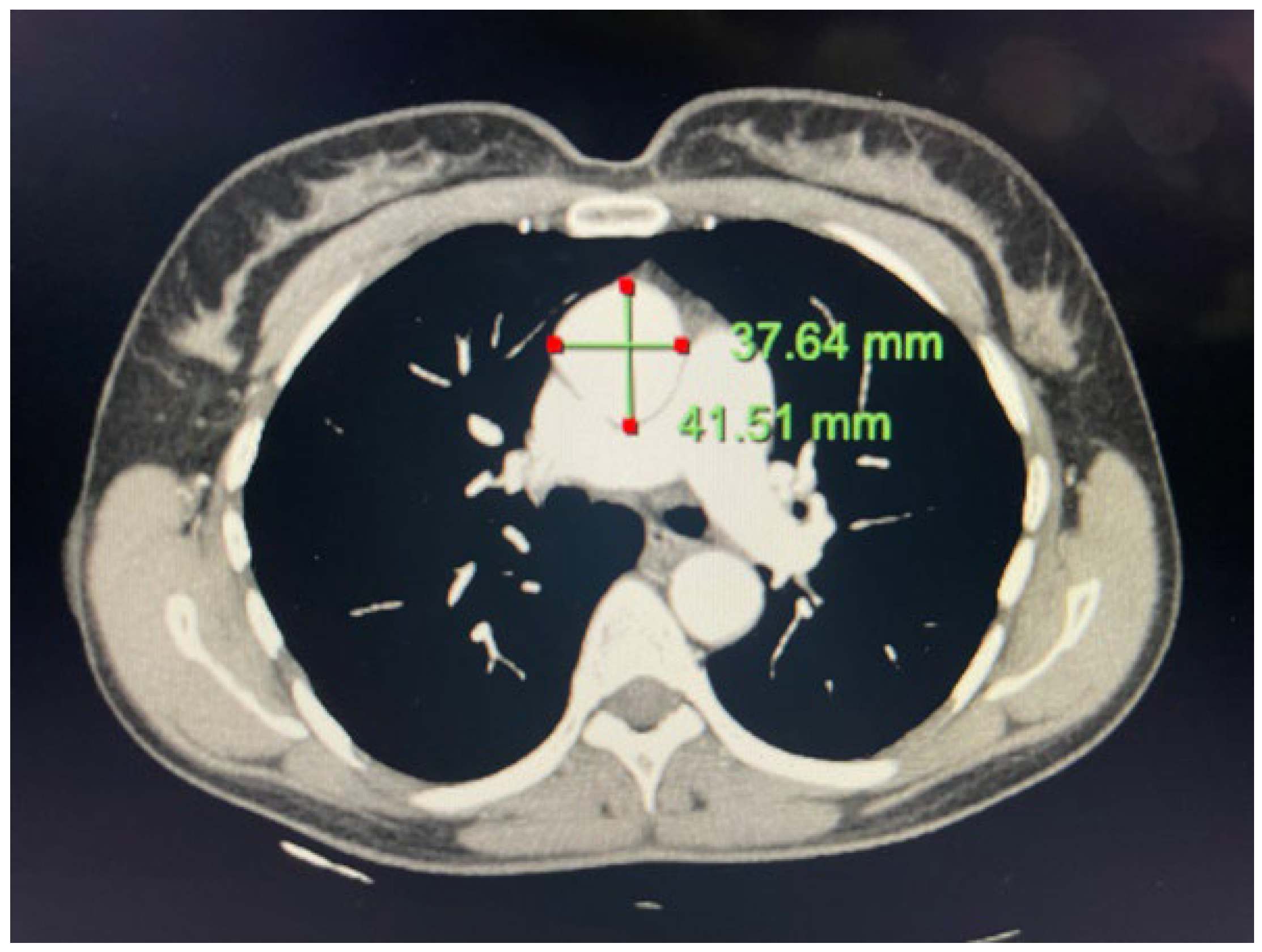
2.2. Case 2
3. Materials and Methods
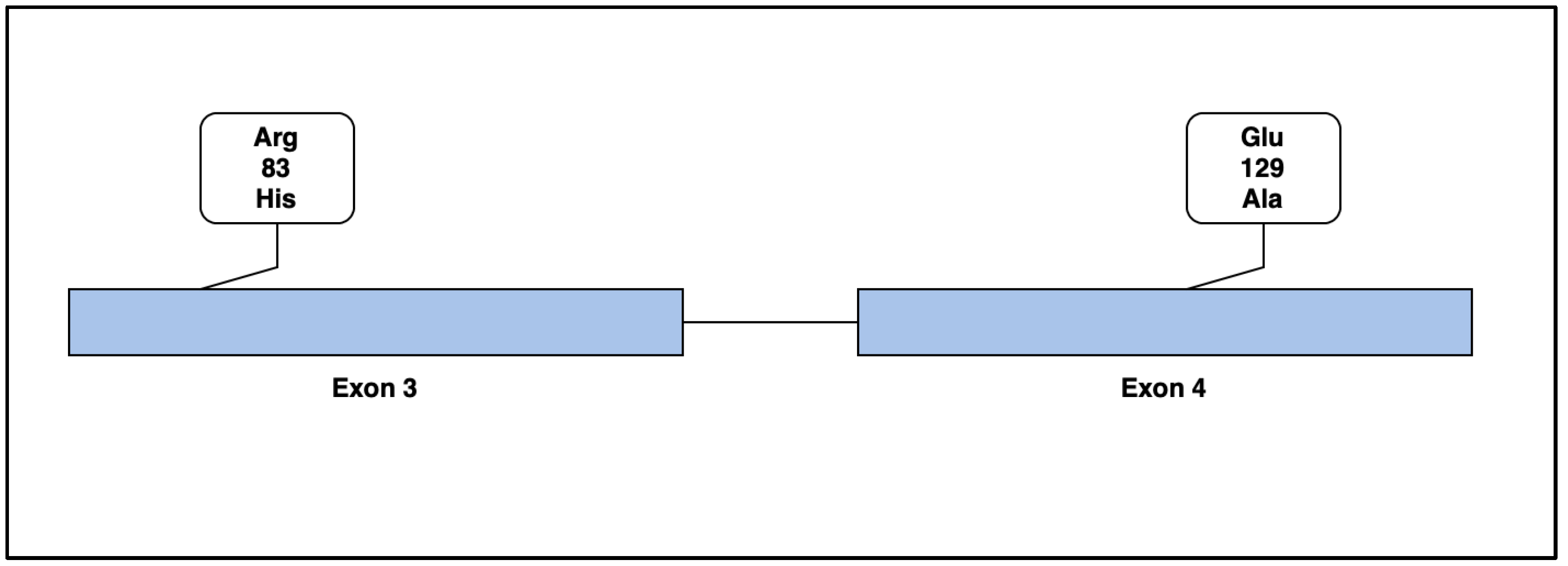
4. Genetic Results
4.1. Case 1
4.2. Case 2
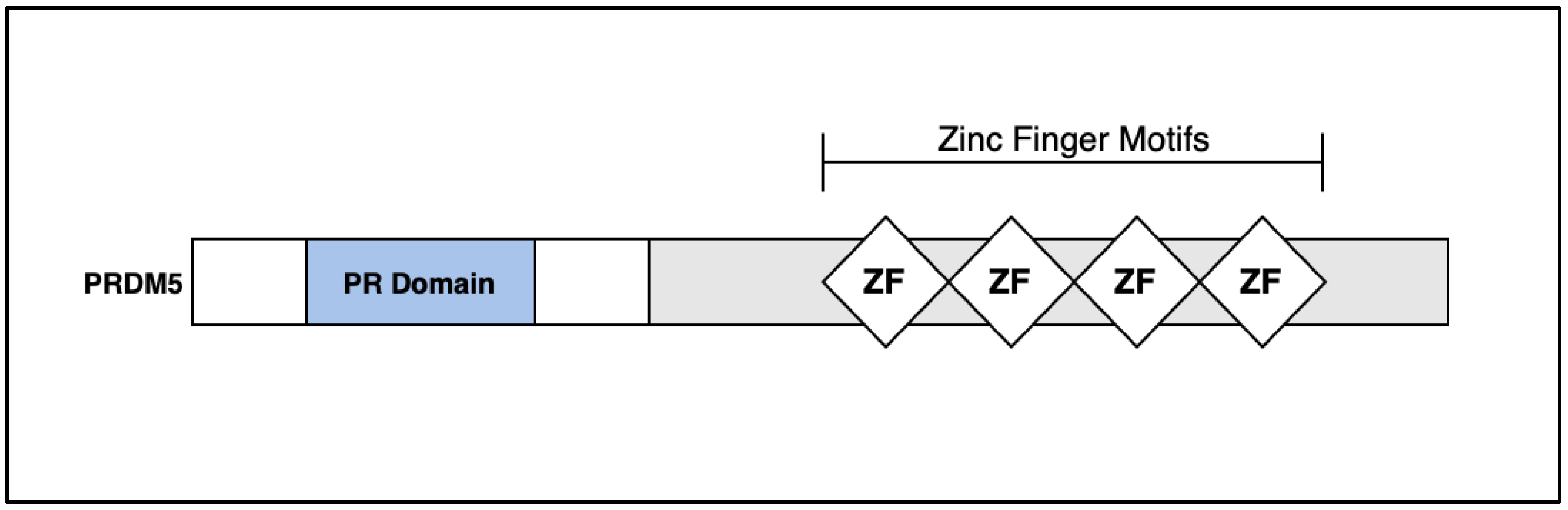
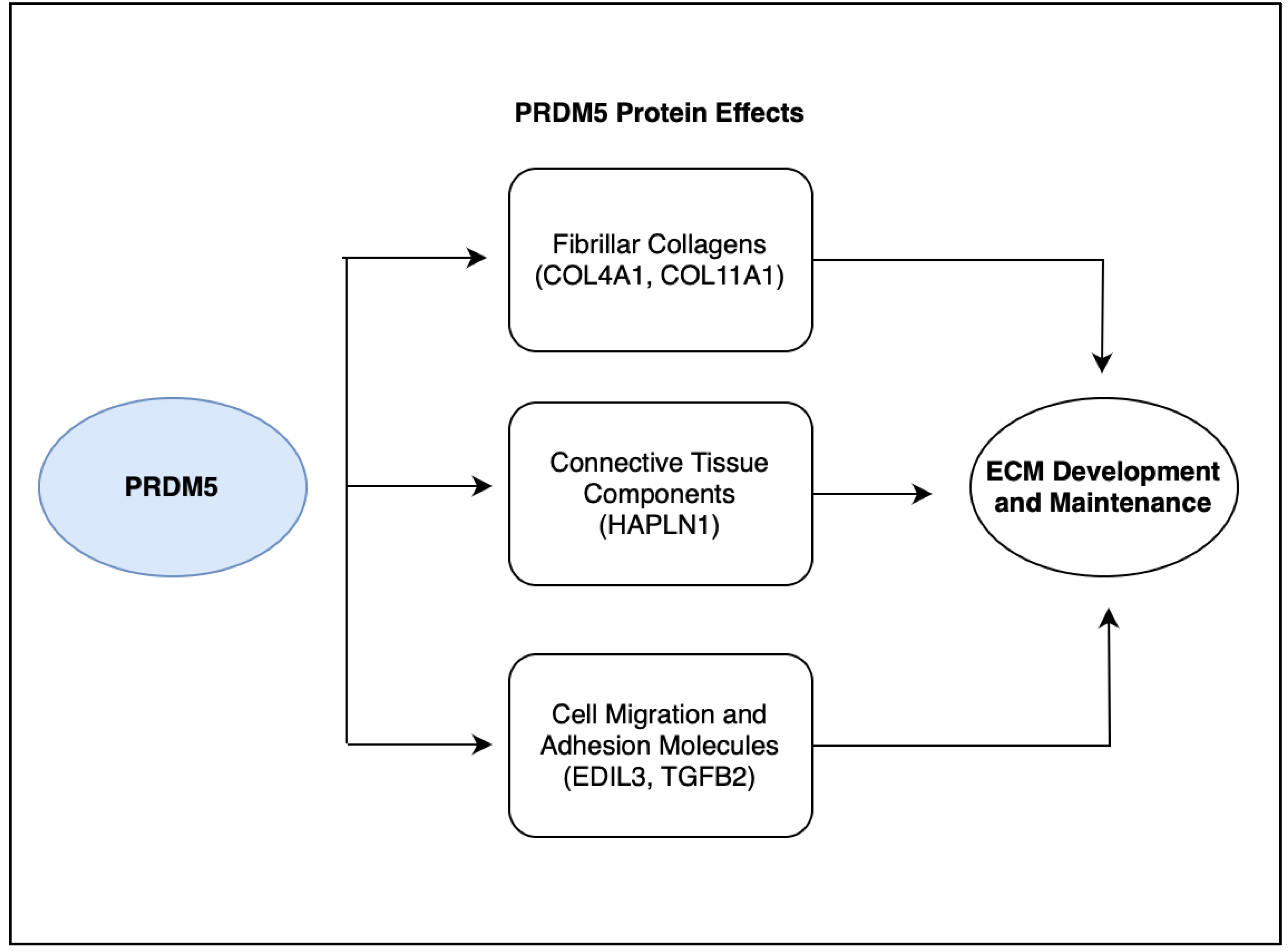
5. Discussion: PRDM5
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senser, E.M.; Misra, S.; Henkin, S. Thoracic Aortic Aneurysm: A Clinical Review. Cardiol. Clin. 2021, 39, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Martin-Blazquez, A.; Heredero, A.; Aldamiz-Echevarria, G.; Martin-Lorenzo, M.; Alvarez-Llamas, G. Non-syndromic thoracic aortic aneurysm: Cellular and molecular insights. J. Pathol. 2021, 254, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Salameh, M.J.; Black, I.J.H.; Ratchford, E.V. Thoracic aortic aneurysm. Vasc. Med. 2018, 23, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Saeyeldin, A.; Zafar, M.A.; Velasquez, C.A.; Ip, K.; Gryaznov, A.; Brownstein, A.J.; Li, Y.; Rizzo, J.A.; Erben, Y.; Ziganshin, B.A.; et al. Natural history of aortic root aneurysms in Marfan syndrome. Ann. Cardiothorac. Surg. 2017, 6, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2006; p. 9. [Google Scholar]
- Antolik, C. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2021; pp. 1–5. [Google Scholar]
- Geng, G. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2022; pp. 1–5. [Google Scholar]
- Kou, E. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2020; pp. 1–5. [Google Scholar]
- Antolik, C. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2022; pp. 1–6. [Google Scholar]
- Geng, J. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections; Ambry Genetics: Aliso Viejo, CA, USA, 2020; pp. 1–5. [Google Scholar]
- Antolik, C. Gene Sequence & Deletion/Duplication Analyses of FBN1 Reflex to TAADNext; Ambry Genetics: Aliso Viejo, CA, USA, 2020; pp. 1–14. [Google Scholar]
- gnomAD. Available online: https://gnomad.broadinstitute.org/gene/ENSG00000138738?dataset=gnomad_r2_1 (accessed on 19 July 2023).
- TAAD Syndrome Genetic Testing | TAADNext | Ambry Genetics. Available online: https://www.ambrygen.com/providers/genetic-testing/12/cardiology/taadnext (accessed on 23 May 2023).
- Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes 2021, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- The yin-yang of PR-domain family genes in tumorigenesis. Histol. Histopathol. 2000, 15, 109–117. [CrossRef]
- PRDM5 PR/SET Domain 5 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/11107 (accessed on 29 May 2023).
- PRDM5 Gene—GeneCards|PRDM5 Protein|PRDM5 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=PRDM5 (accessed on 29 May 2023).
- Dhooge, T.; Van Damme, T.; Syx, D.; Mosquera, L.M.; Nampoothiri, S.; Radhakrishnan, A.; Simsek-Kiper, P.O.; Utine, G.E.; Bonduelle, M.; Migeotte, I.; et al. More than meets the eye: Expanding and reviewing the clinical and mutational spectrum of brittle cornea syndrome. Hum. Mutat. 2021, 42, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.B.; Spencer, H.L.; Daly, S.B.; Manson, F.D.; Zeef, L.A.; Urquhart, J.; Zoppi, N.; Bonshek, R.; Tosounidis, I.; Mohan, M.; et al. Mutations in PRDM5 in Brittle Cornea Syndrome Identify a Pathway Regulating Extracellular Matrix Development and Maintenance. Am. J. Hum. Genet. 2011, 89, 346. [Google Scholar] [CrossRef]
- Porter, L.F.; Gallego-Pinazo, R.; Keeling, C.L.; Kamieniorz, M.; Zoppi, N.; Colombi, M.; Giunta, C.; Bonshek, R.; Manson, F.D.; Black, G.C. Bruch’s membrane abnormalities in PRDM5-related brittle cornea syndrome. Orphanet J. Rare Dis. 2015, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Khimani, F.; Sathyamoorthy, M. Identification of Single-Nucleotide Polymorphisms in ZNF469 in a Patient with Aortoiliac Aneurysmal Disease. Cardiogenetics 2022, 12, 212–217. [Google Scholar] [CrossRef]
- Wolf, A.; Hong, C.; Sathyamoorthy, M. The Novel Phenotype-to-Genotype Association of Novel Single-Nucleotide Variants in Exon 1 and Exon 2 of the Collagen Matrix-Encoding Gene ZNF469 to Arterial Aneurysmal and Dissection Diseases. 2023; Consultants in Cardiovascular Medicine and Science – Fort Worth (CCMS-FW), Fort Worth, TX, USA. manuscript in peer review. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case 1 Amino Acid Characteristics | Arg (R) | His (H) |
|---|---|---|
| Amino Acid Name | Arginine | Histidine |
| Polarity/Charge | Positive Charge | Positive Charge |
| pH | Basic | Basic |
| Residue Weight | 156 | 137 |
| Hydrophobicity Score | −4.5 | −3.2 |
| Hydrophilicity Score | 3 | −0.5 |
| Secondary Structure Propensity | α indifferent/β indifferent | Weak α former/β former |
| Case 2 Amino Acid Characteristics | Glu (E) | Ala (A) |
|---|---|---|
| Amino Acid Name | Glutamic Acid | Alanine |
| Polarity/Charge | Negative Charge | Non-Polar |
| pH | Acidic | Neutral |
| Residue Weight | 129 | 71 |
| Hydrophobicity Score | −3.5 | 1.8 |
| Hydrophilicity Score | 3 | −0.5 |
| Secondary Structure Propensity | Strong α former/strong β breaker | Strong α former/β indifferent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, P.; Wolf, A.; Sathyamoorthy, M. The Association of Novel Single-Nucleotide Variants in the Collagen Matrix-Encoding Gene PRDM5 with Aortic Aneurysmal Disease. Life 2023, 13, 1649. https://doi.org/10.3390/life13081649
Moore P, Wolf A, Sathyamoorthy M. The Association of Novel Single-Nucleotide Variants in the Collagen Matrix-Encoding Gene PRDM5 with Aortic Aneurysmal Disease. Life. 2023; 13(8):1649. https://doi.org/10.3390/life13081649
Chicago/Turabian StyleMoore, Peyton, Adam Wolf, and Mohanakrishnan Sathyamoorthy. 2023. "The Association of Novel Single-Nucleotide Variants in the Collagen Matrix-Encoding Gene PRDM5 with Aortic Aneurysmal Disease" Life 13, no. 8: 1649. https://doi.org/10.3390/life13081649
APA StyleMoore, P., Wolf, A., & Sathyamoorthy, M. (2023). The Association of Novel Single-Nucleotide Variants in the Collagen Matrix-Encoding Gene PRDM5 with Aortic Aneurysmal Disease. Life, 13(8), 1649. https://doi.org/10.3390/life13081649