
The Role of Microorganisms in the Etiopathogenesis of Demyelinating Diseases

Abstract
1. Introduction
2. MS
2.1. EBV
2.2. HERV
2.3. Human Herpes Virus (HHV)
2.4. Gut Microbiota
2.5. Fungi
2.6. Mycobacterium avium Subspecies Paratuberculosis (MAP)
2.7. CMV
3. NMOSD
3.1. Tuberculosis (TB)
3.2. Helicobacter pylori (H. pylori)
3.3. EBV
3.4. HERV
3.5. Gut Microbiota
3.6. Fungi
3.7. Human Immunodeficiency Virus (HIV)
4. MOGAD
5. What about SARS-CoV-2?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gomes, A.B.A.G.R.; Adoni, T. Differential diagnosis of demyelinating diseases: What’s new? Arq. Neuropsiquiatr. 2022, 80 (Suppl. 1), 137–142. [Google Scholar] [CrossRef]
- Ascherio, A. Environmental factors in multiple sclerosis. Expert Rev. Neurother. 2013, 13 (Suppl. 12), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.A.; Tilbery, C.P.; Lana-Peixoto, M.A.; Mendes, M.F.; Kaimen-Maciel, D.R.; Callegaro, D. Historical aspects of multiple sclerosis. Rev. Neurol. 2002, 34, 379–383. [Google Scholar] [PubMed]
- Adams, J.M. Persistent or slow viral infections and related diseases. West J. Med. 1975, 122, 380–393. [Google Scholar]
- Lewy, H.; Rotstein, A.; Kahana, E.; Marrosu, M.G.; Cocco, E.; Laron, Z. Juvenile multiple sclerosis similar to type I diabetes mellitus has a seasonality of month of birth which differs from that in the general population. J. Pediatr. Endocrinol. Metab. 2008, 21, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Dyment, D.A.; Sadovnick, A.D.; Ebers, G.C. Maternal-offspring HLA-DRB1 compatibility in multiple sclerosis. Tissue Antigens 2005, 66, 44–47. [Google Scholar] [CrossRef]
- Laron, Z.; Shulman, L.; Hampe, C.; Blumenfeld, O. Hypothesis: Viral infections of pregnant women may be early triggers of childhood type 1 diabetes and other autoimmune disease. J. Autoimmun. 2023, 135, 102977. [Google Scholar] [CrossRef] [PubMed]
- Pantavou, K.G.; Bagos, P.G. Season of birth and multiple sclerosis: A systematic review and multivariate meta-analysis. J. Neurol. 2020, 267, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Donati, D. Viral infections and multiple sclerosis. Drug Discov. Today Dis. Models 2020, 32, 27–33. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Multiple sclerosis: Autoimmunity and viruses. Curr. Opin. Rheumatol. 2013, 25, 496–501. [Google Scholar] [CrossRef]
- Wu, C.; Jiang, M.L.; Jiang, R.; Pang, T.; Zhang, C.J. The roles of fungus in CNS autoimmune and neurodegeneration disorders. Front. Immunol. 2023, 13, 1077335. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Coghe, G.; Lorefice, L.; Fenu, G.; Cocco, E. Infections and Multiple Sclerosis: From the World to Sardinia, From Sardinia to the World. Front. Immunol. 2021, 12, 728677. [Google Scholar] [CrossRef] [PubMed]
- Cossu, D.; Yokoyama, K.; Nobutaka, N.; Sechi, L.A. From Sardinia to Japan: Update on the role of MAP in multiple sclerosis. Future Microbiol. 2019, 14, 643–646. [Google Scholar]
- Benito-León, J.; Laurence, M. The Role of Fungi in the Etiology of Multiple Sclerosis. Front. Neurol. 2017, 8, 535. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef]
- Nali, L.H.; Olival, G.S.; Montenegro, H.; da Silva, I.T.; Dias-Neto, E.; Naya, H.; Spangenberg, L.; Penalva-de-Oliveira, A.C.; Romano, C.M. Human endogenous retrovirus and multiple sclerosis: A review and transcriptome findings. Mult. Scler. Relat. Disord. 2022, 57, 103383. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Münz, C.; Cohen, J.I.; Ascherio, A. Epstein-Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Steiner, I.; Sriram, S. The “one virus, one disease” model of multiple sclerosis is too constraining. Ann. Neurol. 2007, 62, 529. [Google Scholar] [CrossRef]
- Latifi, T.; Zebardast, A.; Marashi, S.M. The role of human endogenous retroviruses (HERVs) in Multiple Sclerosis and the plausible interplay between HERVs, Epstein-Barr virus infection, and vitamin D. Mult. Scler. Relat. Disord. 2022, 57, 103318. [Google Scholar] [CrossRef]
- Sumaya, C.V.; Myers, L.W.; Ellison, G.W. Epstein-Barr virus antibodies in multiple sclerosis. Arch. Neurol. 1980, 37, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.F.; Bloomer, L.C.; Salmon, V.C.; Bagley, M.H.; Larsen, P.D. Epstein-Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch. Neurol. 1983, 40, 406–408. [Google Scholar] [CrossRef]
- Larsen, P.D.; Bloomer, L.C.; Bray, P.F. Epstein-Barr nuclear antigen and viral capsid antigen antibody titers in multiple sclerosis. Neurology. 1985, 35, 435–438. [Google Scholar] [CrossRef]
- Pakpoor, J.; Ramagopalan, S.V. Epstein-Barr virus is a necessary causative agent in the pathogenesis of multiple sclerosis: Yes. Mult. Scler. 2013, 19, 1690–1691. [Google Scholar] [CrossRef]
- Munger, K.L.; Levin, L.I.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: A prospective study among United States military personnel. Mult. Scler. 2011, 17, 1185–1193. [Google Scholar] [CrossRef]
- Jog, N.R.; McClain, M.T.; Heinlen, L.D.; Gross, T.; Towner, R.; Guthridge, J.M.; Axtell, R.C.; Pardo, G.; Harley, J.B.; James, J.A. Epstein Barr virus nuclear antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J. Autoimmun. 2020, 106, 102332. [Google Scholar] [CrossRef]
- Mameli, G.; Cossu, D.; Cocco, E.; Masala, S.; Frau, J.; Marrosu, M.G.; Sechi, L.A. Epstein-Barr virus and Mycobacterium avium subsp. paratuberculosis peptides are cross recognized by anti-myelin basic protein antibodies in multiple sclerosis patients. J. Neuroimmunol. 2014, 270, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Cepok, S.; Zhou, D.; Srivastava, R.; Nessler, S.; Stei, S.; Büssow, K.; Sommer, N.; Hemmer, B. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J. Clin. Investig. 2005, 115, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kennedy, P.G.; Dupree, C.; Wang, M.; Lee, C.; Pointon, T.; Langford, T.D.; Graner, M.W.; Yu, X. Antibodies from Multiple Sclerosis Brain Identified Epstein-Barr Virus Nuclear Antigen 1 & 2 Epitopes which Are Recognized by Oligoclonal Bands. J. Neuroimmune Pharmacol. 2021, 16, 567–580. [Google Scholar] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- DeLorenze, G.N.; Munger, K.L.; Lennette, E.T.; Orentreich, N.; Vogelman, J.H.; Ascherio, A. Epstein-Barr virus and multiple sclerosis: Evidence of association from a prospective study with long-term follow-up. Arch. Neurol. 2006, 63, 839–844. [Google Scholar] [CrossRef]
- Nielsen, T.R.; Rostgaard, K.; Nielsen, N.M.; Koch-Henriksen, N.; Haahr, S.; Sørensen, P.S.; Hjalgrim, H. Multiple sclerosis after infectious mononucleosis. Arch. Neurol. 2007, 64, 72–75. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 2007, 204, 2899–2912. [Google Scholar] [CrossRef]
- Keane, J.T.; Afrasiabi, A.; Schibeci, S.D.; Swaminathan, S.; Parnell, G.P.; Booth, D.R. The interaction of Epstein-Barr virus encoded transcription factor EBNA2 with multiple sclerosis risk loci is dependent on the risk genotype. EBioMedicine 2021, 71, 103572. [Google Scholar] [CrossRef]
- Menegatti, J.; Schub, D.; Schäfer, M.; Grässer, F.A.; Ruprecht, K. HLA-DRB1*15:01 is a co-receptor for Epstein-Barr virus, linking genetic and environmental risk factors for multiple sclerosis. Eur. J. Immunol. 2021, 51, 2348–2350. [Google Scholar] [CrossRef]
- Sundström, P.; Nyström, M.; Ruuth, K.; Lundgren, E. Antibodies to specific EBNA-1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J. Neuroimmunol. 2009, 215, 102–107. [Google Scholar] [CrossRef]
- Yenamandra, S.P.; Hellman, U.; Kempkes, B.; Darekar, S.D.; Petermann, S.; Sculley, T.; Klein, G.; Kashuba, E. Epstein-Barr virus encoded EBNA-3 binds to vitamin D receptor and blocks activation of its target genes. Cell. Mol. Life Sci. 2010, 67, 4249–4256. [Google Scholar] [CrossRef]
- Farrell, R.A.; Antony, D.; Wall, G.R.; Clark, D.A.; Fisniku, L.; Swanton, J.; Khaleeli, Z.; Schmierer, K.; Miller, D.H.; Giovannoni, G. Humoral immune response to EBV in multiple sclerosis is associated with disease activity on MRI. Neurology 2009, 73, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kvistad, S.; Myhr, K.M.; Holmøy, T.; Bakke, S.; Beiske, A.G.; Bjerve, K.S.; Hovdal, H.; Løken-Amsrud, K.I.; Lilleås, F.; Midgard, R.; et al. Antibodies to Epstein-Barr virus and MRI disease activity in multiple sclerosis. Mult. Scler. 2014, 20, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.P.T.; Gupta, R.; Lindsey, J.W. The cellular immune response against Epstein-Barr virus decreases during ocrelizumab treatment. Mult. Scler. Relat. Disord. 2021, 56, 103282. [Google Scholar] [CrossRef]
- Bilger, A.; Plowshay, J.; Ma, S.; Nawandar, D.; Barlow, E.A.; Romero-Masters, J.C.; Bristol, J.A.; Li, Z.; Tsai, M.H.; Delecluse, H.J.; et al. Leflunomide/teriflunomide inhibit Epstein-Barr virus (EBV)- induced lymphoproliferative disease and lytic viral replication. Oncotarget 2017, 8, 44266–44280. [Google Scholar] [CrossRef]
- Perron, H.; Germi, R.; Bernard, C.; Garcia-Montojo, M.; Deluen, C.; Farinelli, L.; Faucard, R.; Veas, F.; Stefas, I.; Fabriek, B.O.; et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. 2012, 18, 1721–1736. [Google Scholar] [CrossRef]
- van Horssen, J.; van der Pol, S.; Nijland, P.; Amor, S.; Perron, H. Human endogenous retrovirus W in brain lesions: Rationale for targeted therapy in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 8, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Pierquin, J.; Brunel, J.; Gorter, R.; Quétard, C.; Horvat, B.; Amor, S.; Portoukalian, J.; Perron, H. Human Endogenous Retrovirus Type W Envelope from Multiple Sclerosis Demyelinating Lesions Shows Unique Solubility and Antigenic Characteristics. Virol. Sin. 2021, 36, 1006–1026. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.B.; Geny, C.; Deforges, L.; Perron, H.; Tourtelotte, W.; Heltberg, A.; Clausen, J. Expression of endogenous retroviruses in blood mononuclear cells and brain tissue from multiple sclerosis patients. Mult. Scler. 1995, 1, 82–87. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef]
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology 2009, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Laska, M.J.; Brudek, T.; Nissen, K.K.; Christensen, T.; Møller-Larsen, A.; Petersen, T.; Nexø, B.A. Expression of HERV-Fc1, a human endogenous retrovirus, is increased in patients with active multiple sclerosis. J. Virol. 2012, 86, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Joseph, B.; Whittall, T. Potential molecular mimicry between the human endogenous retrovirus W family envelope proteins and myelin proteins in multiple sclerosis. Immunol. Lett. 2017, 183, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Madeira, A.; Burgelin, I.; Perron, H.; Curtin, F.; Lang, A.B.; Faucard, R. MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll-like receptor 4: Relevance of GNbAC1 in multiple sclerosis treatment. J. Neuroimmunol. 2016, 291, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.; Dissing Sørensen, P.; Riemann, H.; Hansen, H.J.; Munch, M.; Haahr, S.; Møller-Larsen, A. Molecular characterization of HERV-H variants associated with multiple sclerosis. Acta Neurol. Scand. 2000, 101, 229–238. [Google Scholar] [CrossRef]
- Mameli, G.; Madeddu, G.; Mei, A.; Uleri, E.; Poddighe, L.; Delogu, L.G.; Maida, I.; Babudieri, S.; Serra, C.; Manetti, R.; et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: The missing link with multiple sclerosis? PLoS ONE 2013, 8, e78474. [Google Scholar] [CrossRef]
- Sutkowski, N.; Conrad, B.; Thorley-Lawson, D.A.; Huber, B.T. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity 2001, 15, 579–589. [Google Scholar] [CrossRef]
- Mostafa, A.; Jalilvand, S.; Shoja, Z.; Nejati, A.; Shahmahmoodi, S.; Sahraian, M.A.; Marashi, S.M. Multiple sclerosis-associated retrovirus, Epstein-Barr virus, and vitamin D status in patients with relapsing remitting multiple sclerosis. J. Med. Virol. 2017, 89, 1309–1313. [Google Scholar] [CrossRef]
- Mameli, G.; Serra, C.; Astone, V.; Castellazzi, M.; Poddighe, L.; Fainardi, E.; Neri, W.; Granieri, E.; Dolei, A. Inhibition of multiple-sclerosis-associated retrovirus as biomarker of interferon therapy. J. Neurovirol. 2008, 14, 73–77. [Google Scholar] [CrossRef]
- Arru, G.; Leoni, S.; Pugliatti, M.; Mei, A.; Serra, C.; Delogu, L.G.; Manetti, R.; Dolei, A.; Sotgiu, S.; Mameli, G. Natalizumab inhibits the expression of human endogenous retroviruses of the W family in multiple sclerosis patients: A longitudinal cohort study. Mult. Scler. 2014, 20, 174–182. [Google Scholar] [CrossRef]
- Derfuss, T.; Curtin, F.; Guebelin, C.; Bridel, C.; Rasenack, M.; Matthey, A.; Du Pasquier, R.; Schluep, M.; Desmeules, J.; Lang, A.B.; et al. A phase IIa randomized clinical study testing GNbAC1, a humanized monoclonal antibody against the envelope protein of multiple sclerosis associated endogenous retrovirus in multiple sclerosis patients-a twelve month follow-up. J. Neuroimmunol. 2015, 285, 68–70. [Google Scholar] [CrossRef]
- Kremer, D.; Förster, M.; Schichel, T.; Göttle, P.; Hartung, H.P.; Perron, H.; Küry, P. The neutralizing antibody GNbAC1 abrogates HERV-W envelope protein-mediated oligodendroglial maturation blockade. Mult. Scler. 2015, 21, 1200–1203. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Pietiläinen-Nicklén, J.; Uotila, L.; Färkkilä, M.; Vaheri, A.; Koskiniemi, M. Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J. Neuroimmunol. 2011, 237, 93–97. [Google Scholar] [CrossRef]
- Keyvani, H.; Zahednasab, H.; Aljanabi, H.A.A.; Asadi, M.; Mirzaei, R.; Esghaei, M.; Karampoor, S. The role of human herpesvirus-6 and inflammatory markers in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2020, 346, 577313. [Google Scholar] [CrossRef]
- Simpson, S., Jr.; Taylor, B.; Dwyer, D.E.; Taylor, J.; Blizzard, L.; Ponsonby, A.L.; Pittas, F.; Dwyer, T.; van der Mei, I. Anti-HHV-6 IgG titer significantly predicts subsequent relapse risk in multiple sclerosis. Mult. Scler. 2012, 18, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Soldan, S.S.; Leist, T.P.; Juhng, K.N.; McFarland, H.F.; Jacobson, S. Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann. Neurol. 2000, 47, 306–313. [Google Scholar] [CrossRef]
- Akhyani, N.; Berti, R.; Brennan, M.B.; Soldan, S.S.; Eaton, J.M.; McFarland, H.F.; Jacobson, S. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: Increased prevalence of HHV-6A in patients with multiple sclerosis. J. Infect. Dis. 2000, 182, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Zang, Y.C.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, E.; Gustafsson, R.; Huang, J.; Biström, M.; Lima Bomfim, I.; Stridh, P.; Khademi, M.; Brenner, N.; Butt, J.; Michel, A.; et al. Increased Serological Response Against Human Herpesvirus 6A Is Associated With Risk for Multiple Sclerosis. Front. Immunol. 2019, 10, 2715. [Google Scholar] [CrossRef]
- Wu, J.; Engdahl, E.; Gustafsson, R.; Fogdell-Hahn, A.; Waterboer, T.; Hillert, J.; Olsson, T.; Alfredsson, L.; Hedström, A.K. High antibody levels against human herpesvirus-6A interact with lifestyle factors in multiple sclerosis development. Mult. Scler. 2022, 28, 383–392. [Google Scholar] [CrossRef]
- Biström, M.; Jons, D.; Engdahl, E.; Gustafsson, R.; Huang, J.; Brenner, N.; Butt, J.; Alonso-Magdalena, L.; Gunnarsson, M.; Vrethem, M.; et al. Epstein-Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur. J. Neurol. 2021, 28, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I.; Zhu, Y.; Ntranos, A.; Clemente, J.C.; Cekanaviciute, E.; Brandstadter, R.; Crabtree-Hartman, E.; Singh, S.; Bencosme, Y.; Debelius, J.; et al. Disease-modifying therapies alter gut microbial composition in MS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 6, e517. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Álvarez, F.; Pérez-Matute, P.; Oteo, J.A.; Marzo-Sola, M.E. The influence of interferon β-1b on gut microbiota composition in patients with multiple sclerosis. Neurologia (Engl. Ed.) 2021, 36, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Maghzi, A.H.; Liu, S.; Tankou, S.K.; Dhang, F.H.; Willocq, V.; Song, A.; Wasén, C.; Tauhid, S.; Chu, R.; et al. Gut Microbiome in Progressive Multiple Sclerosis. Ann. Neurol. 2021, 89, 1195–1211. [Google Scholar] [CrossRef]
- Shah, S.; Locca, A.; Dorsett, Y.; Cantoni, C.; Ghezzi, L.; Lin, Q.; Bokoliya, S.; Panier, H.; Suther, C.; Gormley, M.; et al. Alterations of the gut mycobiome in patients with MS. EBioMedicine 2021, 71, 103557. [Google Scholar] [CrossRef]
- Alonso, R.; Fernández-Fernández, A.M.; Pisa, D.; Carrasco, L. Multiple sclerosis and mixed microbial infections. Direct identification of fungi and bacteria in nervous tissue. Neurobiol. Dis. 2018, 117, 42–61. [Google Scholar] [CrossRef]
- Saroukolaei, S.A.; Ghabaee, M.; Shokri, H.; Badiei, A.; Ghourchian, S. The role of Candida albicans in the severity of multiple sclerosis. Mycoses 2016, 59, 697–704. [Google Scholar] [CrossRef]
- Pisa, D.; Alonso, R.; Jiménez-Jiménez, F.J.; Carrasco, L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 795–801. [Google Scholar] [CrossRef]
- Berg-Hansen, P.; Vandvik, B.; Fagerhol, M.; Holmøy, T. Calprotectin levels in the cerebrospinal fluid reflect disease activity in multiple sclerosis. J. Neuroimmunol. 2009, 216, 98–102. [Google Scholar] [CrossRef]
- Truss, C.O. The role of Candida albicans in human illness. J. Orthomol. Psychiatry 1981, 10, 38. [Google Scholar]
- Cossu, D.; Cocco, E.; Paccagnini, D.; Masala, S.; Ahmed, N.; Frau, J.; Marrosu, M.G.; Sechi, L.A. Association of Mycobacterium avium subsp. paratuberculosis with multiple sclerosis in Sardinian patients. PLoS ONE 2011, 6, e18482. [Google Scholar]
- Frau, J.; Cossu, D.; Coghe, G.; Lorefice, L.; Fenu, G.; Melis, M.; Paccagnini, D.; Sardu, C.; Murru, M.R.; Tranquilli, S.; et al. Mycobacterium avium subsp. paratuberculosis and multiple sclerosis in Sardinian patients: Epidemiology and clinical features. Mult. Scler. 2013, 19, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Cossu, D.; Yokoyama, K.; Sechi, L.A.; Otsubo, S.; Tomizawa, Y.; Momotani, E.; Hattori, N. Humoral response against host-mimetic homologous epitopes of Mycobacterium avium subsp. paratuberculosis in Japanese multiple sclerosis patients. Sci. Rep. 2016, 6, 29227. [Google Scholar] [PubMed]
- Cossu, D.; Masala, S.; Frau, J.; Mameli, G.; Marrosu, M.G.; Cocco, E.; Sechi, L.A. Antigenic epitopes of MAP2694 homologous to T-cell receptor gamma-chain are highly recognized in multiple sclerosis Sardinian patients. Mol. Immunol. 2014, 57, 138–140. [Google Scholar] [CrossRef]
- Cossu, D.; Mameli, G.; Galleri, G.; Cocco, E.; Masala, S.; Frau, J.; Marrosu, M.G.; Manetti, R.; Sechi, L.A. Human interferon regulatory factor 5 homologous epitopes of Epstein-Barr virus and Mycobacterium avium subsp. paratuberculosis induce a specific humoral and cellular immune response in multiple sclerosis patients. Mult. Scler. 2015, 21, 984–995. [Google Scholar] [CrossRef]
- Frau, J.; Cossu, D.; Sardu, C.; Mameli, G.; Coghe, G.; Lorefice, L.; Fenu, G.; Tranquilli, S.; Sechi, L.A.; Marrosu, M.G.; et al. Combining HLA-DRB1-DQB1 and Mycobacterium Avium Subspecies Paratubercolosis (MAP) antibodies in Sardinian multiple sclerosis patients: Associated or independent risk factors? BMC Neurol. 2016, 16, 148. [Google Scholar] [CrossRef]
- Salim, M.A.; Eftekharian, M.M.; Taheri, M.; Yousef Alikhani, M. Determining the IgM and IgG antibody titer against CMV and helicobacter pylori in the serum of multiple sclerosis patients comparing to the control group in Hamadan. Hum. Antibodies 2017, 26, 23–28. [Google Scholar] [CrossRef]
- Grut, V.; Biström, M.; Salzer, J.; Stridh, P.; Jons, D.; Gustafsson, R.; Fogdell-Hahn, A.; Huang, J.; Brenner, N.; Butt, J.; et al. Cytomegalovirus seropositivity is associated with reduced risk of multiple sclerosis-a presymptomatic case-control study. Eur. J. Neurol. 2021, 28, 3072–3079. [Google Scholar] [CrossRef]
- Sundqvist, E.; Bergström, T.; Daialhosein, H.; Nyström, M.; Sundström, P.; Hillert, J.; Alfredsson, L.; Kockum, I.; Olsson, T. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult. Scler. 2014, 20, 165–173. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef]
- Lassmann, H.; Niedobitek, G.; Aloisi, F.; Middeldorp, J.M.; NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: A controversial issue--report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain 2011, 134 Pt 9, 2772–2786. [Google Scholar] [CrossRef] [PubMed]
- Makhani, N.; Tremlett, H. The multiple sclerosis prodrome. Nat. Rev. Neurol. 2021, 17, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Laurence, M.; Benito-León, J. Epstein-Barr virus and multiple sclerosis: Updating Pender’s hypothesis. Mult. Scler. Relat. Disord. 2017, 16, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Jakimovski, D.; Ramanathan, M.; Weinstock-Guttman, B.; Zivadinov, R. The role of Epstein-Barr virus in multiple sclerosis: From molecular pathophysiology to in vivo imaging. Neural. Regen. Res. 2019, 14, 373–386. [Google Scholar]
- Serafini, B.; Rosicarelli, B.; Veroni, C.; Mazzola, G.A.; Aloisi, F. Epstein-Barr Virus-Specific CD8 T Cells Selectively Infiltrate the Brain in Multiple Sclerosis and Interact Locally with Virus-Infected Cells: Clue for a Virus-Driven Immunopathological Mechanism. J. Virol. 2019, 93, e00980-19. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Hochberg, D.; Babcock, G.J.; Freeman, R.B.; Thorley-Lawson, D.A. The dispersal of mucosal memory B cells: Evidence from persistent EBV infection. Immunity 2002, 16, 745–754. [Google Scholar] [CrossRef]
- Sedighi, S.; Gholizadeh, O.; Yasamineh, S.; Akbarzadeh, S.; Amini, P.; Favakehi, P.; Afkhami, H.; Firouzi-Amandi, A.; Pahlevan, D.; Eslami, M.; et al. Comprehensive Investigations Relationship Between Viral Infections and Multiple Sclerosis Pathogenesis. Curr. Microbiol. 2022, 80, 15. [Google Scholar] [CrossRef]
- Dolei, A.; Perron, H. The multiple sclerosis-associated retrovirus and its HERV-W endogenous family: A biological interface between virology, genetics, and immunology in human physiology and disease. J. Neurovirol. 2009, 15, 4–13. [Google Scholar] [CrossRef]
- Aloisi, F.; Cross, A.H. MINI-review of Epstein-Barr virus involvement in multiple sclerosis etiology and pathogenesis. J. Neuroimmunol. 2022, 371, 577935. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Giovannoni, G.; Cuzick, J.; Dobson, R. Systematic review and meta-analysis of the association between Epstein-Barr virus, multiple sclerosis and other risk factors. Mult. Scler. 2020, 26, 1281–1297. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Hernandez, O.D.; Martínez-Cáceres, E.M.; Presas-Rodríguez, S.; Ramo-Tello, C. Epstein-Barr Virus and Multiple Sclerosis: A Convoluted Interaction and the Opportunity to Unravel Predictive Biomarkers. Int. J. Mol. Sci. 2023, 24, 7407. [Google Scholar] [CrossRef]
- Ruprecht, K. The role of Epstein-Barr virus in the etiology of multiple sclerosis: A current review. Expert Rev. Clin. Immunol. 2020, 16, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Kosowicz, J.G.; Lee, J.; Peiffer, B.; Guo, Z.; Chen, J.; Liao, G.; Hayward, S.D.; Liu, J.O.; Ambinder, R.F. Drug Modulators of B Cell Signaling Pathways and Epstein-Barr Virus Lytic Activation. J. Virol. 2017, 91, e00747-17. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Giovannoni, G.; Salvetti, M. Epstein-Barr virus as a cause of multiple sclerosis: Opportunities for prevention and therapy. Lancet Neurol. 2023, 22, 338–349. [Google Scholar] [CrossRef]
- Wei, C.J.; Bu, W.; Nguyen, L.A.; Batchelor, J.D.; Kim, J.; Pittaluga, S.; Fuller, J.R.; Nguyen, H.; Chou, T.H.; Cohen, J.I.; et al. A bivalent Epstein-Barr virus vaccine induces neutralizing antibodies that block infection and confer immunity in humanized mice. Sci. Transl. Med. 2022, 14, eabf3685. [Google Scholar] [CrossRef]
- Perron, H.; Bernard, C.; Bertrand, J.B.; Lang, A.B.; Popa, I.; Sanhadji, K.; Portoukalian, J. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J. Neurol. Sci. 2009, 286, 65–72. [Google Scholar] [CrossRef]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef]
- Arneth, B. Up-to-date knowledge about the association between multiple sclerosis and the reactivation of human endogenous retrovirus infections. J. Neurol. 2018, 265, 1733–1739. [Google Scholar] [CrossRef]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef]
- Bogdanos, D.P.; Smyk, D.S.; Invernizzi, P.; Rigopoulou, E.I.; Blank, M.; Pouria, S.; Shoenfeld, Y. Infectome: A platform to trace infectious triggers of autoimmunity. Autoimmun. Rev. 2013, 12, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Ørskov, A.D.; Bjerregaard, A.M.; Unnikrishnan, A.; Holmberg-Thydén, S.; Borch, A.; Jensen, K.V.; Anande, G.; Bentzen, A.K.; Marquard, A.M.; et al. Human endogenous retroviruses form a reservoir of T cell targets in hematological cancers. Nat. Commun. 2020, 11, 5660. [Google Scholar] [CrossRef] [PubMed]
- Brütting, C.; Stangl, G.I.; Staege, M.S. Vitamin D, Epstein-Barr virus, and endogenous retroviruses in multiple sclerosis-facts and hypotheses. J. Integr. Neurosci. 2021, 20, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Dolei, A. The aliens inside us: HERV-W endogenous retroviruses and multiple sclerosis. Mult. Scler. 2018, 24, 42–47. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.J.; Yang, L.; Yang, C.S.; Yi, M.; Zhang, S.N.; Wang, N.; Huang, C.N.; Liu, M.Q. Positive association of herpes simplex virus-IgG with multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2021, 47, 102633. [Google Scholar] [CrossRef]
- Ablashi, D.; Agut, H.; Alvarez-Lafuente, R.; Clark, D.A.; Dewhurst, S.; DiLuca, D.; Flamand, L.; Frenkel, N.; Gallo, R.; Gompels, U.A.; et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch. Virol. 2014, 159, 863–870. [Google Scholar] [CrossRef]
- Merelli, E.; Bedin, R.; Sola, P.; Barozzi, P.; Mancardi, G.L.; Ficarra, G.; Franchini, G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J. Neurol. 1997, 244, 450–454. [Google Scholar] [CrossRef]
- Voumvourakis, K.I.; Kitsos, D.K.; Tsiodras, S.; Petrikkos, G.; Stamboulis, E. Human herpesvirus 6 infection as a trigger of multiple sclerosis. Mayo. Clin. Proc. 2010, 85, 1023–1030. [Google Scholar] [CrossRef]
- Lundström, W.; Gustafsson, R. Human Herpesvirus 6A Is a Risk Factor for Multiple Sclerosis. Front. Immunol. 2022, 13, 840753. [Google Scholar] [CrossRef]
- Fierz, W. Multiple sclerosis: An example of pathogenic viral interaction? Virol. J. 2017, 14, 42. [Google Scholar] [CrossRef]
- Dunn, N.; Kharlamova, N.; Fogdell-Hahn, A. The role of herpesvirus 6A and 6B in multiple sclerosis and epilepsy. Scand. J. Immunol. 2020, 92, e12984. [Google Scholar] [CrossRef]
- Hube, B. From commensal to pathogen: Stage- and tissue-specific gene expression of Candida albicans. Curr. Opin. Microbiol. 2004, 7, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Hohlfeld, R.; Baranzini, S.E. The role of the gut microbiota in multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Cronin, O.; O’Sullivan, O.; Barton, W.; Cotter, P.D.; Molloy, M.G.; Shanahan, F. Gut microbiota: Implications for sports and exercise medicine. Br. J. Sports Med. 2017, 51, 700–701. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Ye, Y.; Wang, F.; Chen, F.; Zheng, C. The Role of Microbiota in Infant Health: From Early Life to Adulthood. Front. Immunol. 2021, 12, 708472. [Google Scholar] [CrossRef] [PubMed]
- Altieri, C.; Speranza, B.; Corbo, M.R.; Sinigaglia, M.; Bevilacqua, A. Gut-Microbiota, and Multiple Sclerosis: Background, Evidence, and Perspectives. Nutrients 2023, 15, 942. [Google Scholar] [CrossRef]
- Miyauchi, E.; Shimokawa, C.; Steimle, A.; Desai, M.S.; Ohno, H. The impact of the gut microbiome on extra-intestinal autoimmune diseases. Nat. Rev. Immunol. 2023, 23, 9–23. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Waubant, E.; Chehoud, C.; Kuczynski, J.; DeSantis, T.Z.; Warrington, J.; Venkatesan, A.; Fraser, C.M.; Mowry, E.M. Gut microbiota in multiple sclerosis: Possible influence of immunomodulators. J. Investig. Med. 2015, 63, 729–734. [Google Scholar] [CrossRef]
- Pilotto, S.; Zoledziewska, M.; Fenu, G.; Cocco, E.; Lorefice, L. Disease-modifying therapy for multiple sclerosis: Implications for gut microbiota. Mult. Scler. Relat. Disord. 2023, 73, 104671. [Google Scholar] [CrossRef] [PubMed]
- Severance, E.G.; Gressitt, K.L.; Stallings, C.R.; Katsafanas, E.; Schweinfurth, L.A.; Savage, C.L.; Adamos, M.B.; Sweeney, K.M.; Origoni, A.E.; Khushalani, S.; et al. Candida albicans exposures, sex specificity and cognitive deficits in schizophrenia and bipolar disorder. NPJ Schizophr. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Purzycki, C.B.; Shain, D.H. Fungal toxins and multiple sclerosis: A compelling connection. Brain Res. Bull. 2010, 82, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Akgün, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun. Rev. 2020, 19, 102647. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Santos, N.; Gaffen, S.L. Th17 cells in immunity to Candida albicans. Cell Host Microbe 2012, 11, 425–435. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Marijnissen, R.J.; Kullberg, B.J.; Koenen, H.J.; Cheng, S.C.; Joosten, I.; van den Berg, W.B.; Williams, D.L.; van der Meer, J.W.; Joosten, L.A.; et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009, 5, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Marijnissen, R.J.; Koenders, M.I.; van de Veerdonk, F.L.; Dulos, J.; Netea, M.G.; Boots, A.M.; Joosten, L.A.; van den Berg, W.B. Exposure to Candida albicans polarizes a T-cell driven arthritis model towards Th17 responses, resulting in a more destructive arthritis. PLoS ONE 2012, 7, e38889. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Cocco, E.; Frau, J.; Marrosu, M.G.; Sechi, L.A. Epstein Barr Virus and Mycobacterium avium subsp. paratuberculosis peptides are recognized in sera and cerebrospinal fluid of MS patients. Sci. Rep. 2016, 6, 22401. [Google Scholar] [CrossRef]
- Dow, C.T. Proposing BCG Vaccination for Mycobacterium avium ss. paratuberculosis (MAP) Associated Autoimmune Diseases. Microorganisms 2020, 8, 212. [Google Scholar] [CrossRef]
- Amato, M.P.; De Stefano, N.; Inglese, M.; Morena, E.; Ristori, G.; Salvetti, M.; Trojano, M. Secondary Prevention in Radiologically Isolated Syndromes and Prodromal Stages of Multiple Sclerosis. Front. Neurol. 2022, 13, 787160. [Google Scholar] [CrossRef]
- Ristori, G.; Buzzi, M.G.; Sabatini, U.; Giugni, E.; Bastianello, S.; Viselli, F.; Buttinelli, C.; Ruggieri, S.; Colonnese, C.; Pozzilli, C.; et al. Use of Bacille Calmette-Guèrin (BCG) in multiple sclerosis. Neurology 1999, 53, 1588–1589. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, A.; Buzzi, M.G.; Giugni, E.; Sabatini, U.; Bastianello, S.; Pozzilli, C.; Salvetti, M.; Ristori, G. The effect of Bacille Calmette-Guérin on the evolution of new enhancing lesions to hypointense T1 lesions in relapsing remitting MS. J. Neurol. 2003, 250, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Ristori, G.; Romano, S.; Cannoni, S.; Visconti, A.; Tinelli, E.; Mendozzi, L.; Cecconi, P.; Lanzillo, R.; Quarantelli, M.; Buttinelli, C.; et al. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology 2014, 82, 41–48. [Google Scholar] [CrossRef]
- Baasch, S.; Ruzsics, Z.; Henneke, P. Cytomegaloviruses and Macrophages-Friends and Foes From Early on? Front. Immunol. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Liu, C.; Wang, G.; Liu, H.; Li, Y.; Li, J.; Dai, Y.; Hu, X. CD226 Gly307Ser association with neuromyelitis optica in Southern Han Chinese. Can. J. Neurol. Sci. 2012, 39, 488–490. [Google Scholar] [CrossRef]
- Zhong, X.; Zhou, Y.; Lu, T.; Wang, Z.; Fang, L.; Peng, L.; Kermode, A.G.; Qiu, W. Infections in neuromyelitis optica spectrum disorder. J. Clin. Neurosci. 2018, 47, 14–19. [Google Scholar] [CrossRef]
- Barros, P.O.; Linhares, U.C.; Teixeira, B.; Kasahara, T.M.; Ferreira, T.B.; Alvarenga, R.; Hygino, J.; Silva-Filho, R.G.; Bittencourt, V.C.; Andrade, R.M.; et al. High in vitro immune reactivity to Escherichia coli in neuromyelitis optica patients is correlated with both neurological disabilities and elevated plasma lipopolysaccharide levels. Hum. Immunol. 2013, 74, 1080–1087. [Google Scholar] [CrossRef]
- Koga, M.; Takahashi, T.; Kawai, M.; Fujihara, K.; Kanda, T. A serological analysis of viral and bacterial infections associated with neuromyelitis optica. J. Neurol. Sci. 2011, 300, 19–22. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, B.G. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar] [CrossRef]
- Bergamaschi, R.; Ghezzi, A. Devic’s neuromyelitis optica: Clinical features and prognostic factors. Neurol. Sci. 2004, 25 (Suppl. 4), S364–S367. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Hemmer, B.; Mühlau, M. The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J. Autoimmun. 2010, 34, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Münz, C.; Lünemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Graves, J.; Grandhe, S.; Weinfurtner, K.; Krupp, L.; Belman, A.; Chitnis, T.; Ness, J.; Weinstock-Guttman, B.; Gorman, M.; Patterson, M. Protective environmental factors for neuromyelitis optica. Neurology 2014, 83, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Brey, N.; Henning, F. Relapsing neuromyelitis optica temporally related to recurrent pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 2014, 18, 632–633. [Google Scholar] [CrossRef]
- Li, R.; Zhong, X.; Qiu, W.; Wu, A.; Dai, Y.; Lu, Z.; Hu, X. Association between neuromyelitis optica and tuberculosis in a Chinese population. BMC Neurol. 2014, 14, 33. [Google Scholar] [CrossRef]
- Zatjirua, V.; Butler, J.; Carr, J.; Henning, F. Neuromyelitis optica and pulmonary tuberculosis: A case-control study. Int. J. Tuberc. Lung Dis. 2011, 15, 1675–1680. [Google Scholar] [CrossRef]
- Rafai, M.A.; Boulaajaj, F.Z.; Gynerane, M.; El Moutawakkil, B.; Slassi, I. Devic-like syndrome in the course of pulmonary tuberculosis. Acta Neurol. Belg. 2010, 110, 196–200. [Google Scholar]
- Feng, Y.Q.; Guo, N.; Huang, F.; Chen, X.; Sun, Q.S.; Liu, J.X. Anti-tuberculosis treatment for Devic’s neuromyelitis optica. J. Clin. Neurosci. 2010, 17, 1372–1377. [Google Scholar] [CrossRef]
- Papais-Alvarenga, R.M.; Miranda-Santos, C.M.; Puccioni-Sohler, M.; de Almeida, A.M.; Oliveira, S.; Basilio De Oliveira, C.A.; Alvarenga, H.; Poser, C.M. Optic neuromyelitis syndrome in Brazilian patients. J. Neurol. Neurosurg. Psychiatry 2002, 73, 429–435. [Google Scholar] [CrossRef]
- Yoshimura, S.; Isobe, N.; Matsushita, T.; Yonekawa, T.; Masaki, K.; Sato, S.; Kawano, Y.; Kira, J.; South Japan Multiple Sclerosis Genetics Consortium. Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti-aquaporin 4 antibody status. J. Neurol. Neurosurg. Psychiatry 2013, 84, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Gao, C.; Qiu, W.; Hu, X.; Shu, Y.; Peng, F.; Lu, Z. Helicobacter pylori infection in Neuromyelitis Optica and Multiple Sclerosis. Neuroimmunomodulation 2013, 20, 107–112. [Google Scholar] [CrossRef]
- Li, W.; Minohara, M.; Piao, H.; Matsushita, T.; Masaki, K.; Matsuoka, T.; Isobe, N.; Su, J.J.; Ohyagi, Y.; Kira, J. Association of anti-Helicobacter pylori neutrophil-activating protein antibody response with anti-aquaporin-4 autoimmunity in Japanese patients with multiple sclerosis and neuromyelitis optica. Mult. Scler. 2009, 15, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Barzegar, M.; Nouri, H.; Mirmosayyeb, O.; Motedayyen, H.; Nehzat, N.; Shaygannejad, V. Association Between Helicobacter Pylori Infection and Seronegative Neuromyelitis Optica Spectrum Disorder. Casp. J. Neurol. Sci. 2023, 9, 9–14. [Google Scholar] [CrossRef]
- Masuda, S.; Mori, M.; Arai, K.; Uzawa, A.; Muto, M.; Uchida, T.; Masuda, H.; Kuwabara, S. Epstein-Barr virus persistence and reactivation in neuromyelitis optica. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Mori, M. Association between Multiple Sclerosis or Neuromyelitis Optica and Epstein-Barr Virus. Brain Nerve 2015, 67, 881–890. [Google Scholar]
- Arru, G.; Sechi, E.; Mariotto, S.; Farinazzo, A.; Mancinelli, C.; Alberti, D.; Ferrari, S.; Gajofatto, A.; Capra, R.; Monaco, S.; et al. Antibody response against HERV-W env surface peptides differentiates multiple sclerosis and neuromyelitis optica spectrum disorder. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317742425. [Google Scholar] [CrossRef]
- Arru, G.; Sechi, E.; Mariotto, S.; Zarbo, I.R.; Ferrari, S.; Gajofatto, A.; Monaco, S.; Deiana, G.A.; Bo, M.; Sechi, L.A.; et al. Antibody response against HERV-W in patients with MOG-IgG associated disorders, multiple sclerosis and NMOSD. J. Neuroimmunol. 2020, 338, 577110. [Google Scholar] [CrossRef]
- Cree, B.A.; Spencer, C.M.; Varrin-Doyer, M.; Baranzini, S.E.; Zamvil, S.S. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann. Neurol. 2016, 80, 443–447. [Google Scholar] [CrossRef]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.; Zamvil, S.S. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef]
- Mathew, T.; Avati, A.; D’Souza, D.; Therambil, M.; Baptist, A.A.; Shaji, A.; Nadig, R.; Rockey, S.M.; Parry, G. HIV infection associated neuromyelitis optica spectrum disorder: Clinical features, imaging findings, management and outcomes. Mult. Scler. Relat. Disord. 2019, 27, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Younas, M.; Psomas, C.; Reynes, J.; Corbeau, P. Immune activation in the course of HIV-1 infection: Causes, phenotypes and persistence under therapy. HIV Med. 2016, 17, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, A.M.; Singh, P.; Smith, R.G. Neuromyelitis optica in patients with coexisting human immunodeficiency virus infections. Mult. Scler. 2013, 19, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Zavos, C.; Polyzos, S.A.; Michael, S.; Tsiaousi, E.; Vardaka, E.; Katsinelos, P.; Kouklakis, G.; Paikos, D.; Gavalas, E.; et al. Relationship between Helicobacter pylori infection and autoimmune disorders. Clin. Chem. Lab. Med. 2013, 51, e73–e74. [Google Scholar] [CrossRef] [PubMed]
- Lederman, M.M.; Funderburg, N.T.; Sekaly, R.P.; Klatt, N.R.; Hunt, P.W. Residual immune dysregulation syndrome in treated HIV infection. Adv. Immunol. 2013, 119, 51–83. [Google Scholar]
- Delgado, S.R.; Maldonado, J.; Rammohan, K.W. CNS demyelinating disorder with mixed features of neuromyelitis optica and multiple sclerosis in HIV-1 infection. Case report and literature review. J. Neurovirol. 2014, 20, 531–537. [Google Scholar] [CrossRef]
- López-Chiriboga, A.S.; Majed, M.; Fryer, J.; Dubey, D.; McKeon, A.; Flanagan, E.P.; Jitprapaikulsan, J.; Kothapalli, N.; Tillema, J.M.; Chen, J.; et al. Association of MOG-IgG Serostatus With Relapse After Acute Disseminated Encephalomyelitis and Proposed Diagnostic Criteria for MOG-IgG-Associated Disorders. JAMA Neurol. 2018, 75, 1355–1363. [Google Scholar] [CrossRef]
- Ramanathan, S.; Mohammad, S.; Tantsis, E.; Nguyen, T.K.; Merheb, V.; Fung, V.S.C.; White, O.B.; Broadley, S.; Lechner-Scott, J.; Vucic, S.; et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J. Neurol. Neurosurg. Psychiatry 2018, 89, 127–137. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Messina, S.; Woodhall, M.R.; Raza, N.; Everett, R.; Roca-Fernandez, A.; Tackley, G.; Hamid, S.; Sheard, A.; Reynolds, G.; et al. Clinical presentation and prognosis in MOG-antibody disease: A UK study. Brain 2017, 140, 3128–3138. [Google Scholar] [CrossRef]
- Sechi, E.; Buciuc, M.; Pittock, S.J.; Chen, J.J.; Fryer, J.P.; Jenkins, S.M.; Budhram, A.; Weinshenker, B.G.; Lopez-Chiriboga, A.S.; Tillema, J.M.; et al. Positive Predictive Value of Myelin Oligodendrocyte Glycoprotein Autoantibody Testing. JAMA Neurol. 2021, 78, 741–746. [Google Scholar] [CrossRef]
- Esposito, S.; Di Pietro, G.M.; Madini, B.; Mastrolia, M.V.; Rigante, D. A spectrum of inflammation and demyelination in acute disseminated encephalomyelitis (ADEM) of children. Autoimmun. Rev. 2015, 14, 923–929. [Google Scholar] [CrossRef]
- Thomas, G.S.; Hussain, I.H. Acute disseminated encephalomyelitis: A report of six cases. Med. J. Malays. 2004, 59, 342–351. [Google Scholar]
- Chowdhary, J.; Ashraf, S.M.; Khajuria, K. Measles with acute disseminated encephalomyelitis (ADEM). Indian Pediatr. 2009, 46, 72–74. [Google Scholar] [PubMed]
- Ozkale, Y.; Erol, I.; Ozkale, M.; Demir, S.; Alehan, F. Acute disseminated encephalomyelitis associated with influenza A H1N1 infection. Pediatr. Neurol. 2012, 47, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, P.; Batocchi, A.P.; Colosimo, C.; Cattani, P.; Stefanini, M.C.; Colitto, F.; Tonali, P.; Guzzetta, F. Positive PCR for enterovirus in the cerebrospinal fluid of a child with acute disseminated encephalomyelitis. J. Neurol. 2004, 251, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.A.; Collins, A.; Cohen, M.E.; Duffner, P.K.; Faden, H. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 2004, 113, e73–e76. [Google Scholar] [PubMed]
- Nakamura, Y.; Nakajima, H.; Tani, H.; Hosokawa, T.; Ishida, S.; Kimura, F.; Kaneko, K.; Takahashi, T.; Nakashima, I. Anti-MOG antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: A case report. BMC Neurol. 2017, 17, 76. [Google Scholar] [CrossRef]
- Nakamura, M.; Iwasaki, Y.; Takahashi, T.; Kaneko, K.; Nakashima, I.; Kunieda, T.; Kaneko, S.; Kusaka, H. A case of MOG antibody-positive bilateral optic neuritis and meningoganglionitis following a genital herpes simplex virus infection. Mult. Scler. Relat. Disord. 2017, 17, 148–150. [Google Scholar] [CrossRef]
- Bonagiri, P.; Park, D.; Ingebritsen, J.; Christie, L.J. Seropositive anti-MOG antibody-associated acute disseminated encephalomyelitis (ADEM): A sequelae of Mycoplasma pneumoniae infection. BMJ Case Rep. 2020, 13, e234565. [Google Scholar] [CrossRef]
- Huang, X.; Guo, R.; Li, C.; Long, X.; Yang, T.; Hou, X.; Wei, X.; Ou, M. A case of anti-myelin oligodendrocyte glycoprotein (MOG)-immunoglobulin G (IgG) associated disorder (MOGAD) with clinical manifestations of acute disseminated encephalomyelitis: Secondary to mycoplasma pneumoniae infection. Heliyon 2023, 9, e13470. [Google Scholar] [CrossRef]
- Huda, S.; Whittam, D.; Jackson, R.; Karthikeayan, V.; Kelly, P.; Linaker, S.; Mutch, K.; Kneen, R.; Woodhall, M.; Murray, K.; et al. Predictors of relapse in MOG antibody associated disease: A cohort study. BMJ Open 2021, 11, e055392. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.; Rahmlow, M. Myelin oligodendrocyte glycoprotein associated transverse myelitis following brain abscess: Case report and literature review. J. Neuroimmunol. 2022, 372, 577967. [Google Scholar] [CrossRef] [PubMed]
- de Luca, V.; Martins Higa, A.; Malta Romano, C.; Pimenta Mambrini, G.; Peroni, L.A.; Trivinho-Strixino, F.; Lima Leite, F. Cross-reactivity between myelin oligodendrocyte glycoprotein and human endogenous retrovirus W protein: Nanotechnological evidence for the potential trigger of multiple sclerosis. Micron 2019, 120, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Abud, A.M.; Rizvi, S.; Zainah, H.; O’Brien, T.C.; Villamar, M.F. Aggressive MOGAD with bilateral corticospinal tract lesions following infection with Jamestown Canyon virus. J. Neuroimmunol. 2022, 373, 577997. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Olivé-Cirera, G.; Martínez-Hernandez, E.; Sepulveda, M.; Ruiz-Garcia, R.; Muñoz-Batista, M.; Ariño, H.; González-Álvarez, V.; Felipe-Rucián, A.; Jesús Martínez-González, M.; et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: A multicentre observational study. Lancet Neurol. 2020, 19, 234–246. [Google Scholar] [CrossRef]
- Lotan, I.; Nishiyama, S.; Manzano, G.S.; Lydston, M.; Levy, M. COVID-19 and the risk of CNS demyelinating diseases: A systematic review. Front. Neurol. 2022, 13, 970383. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]



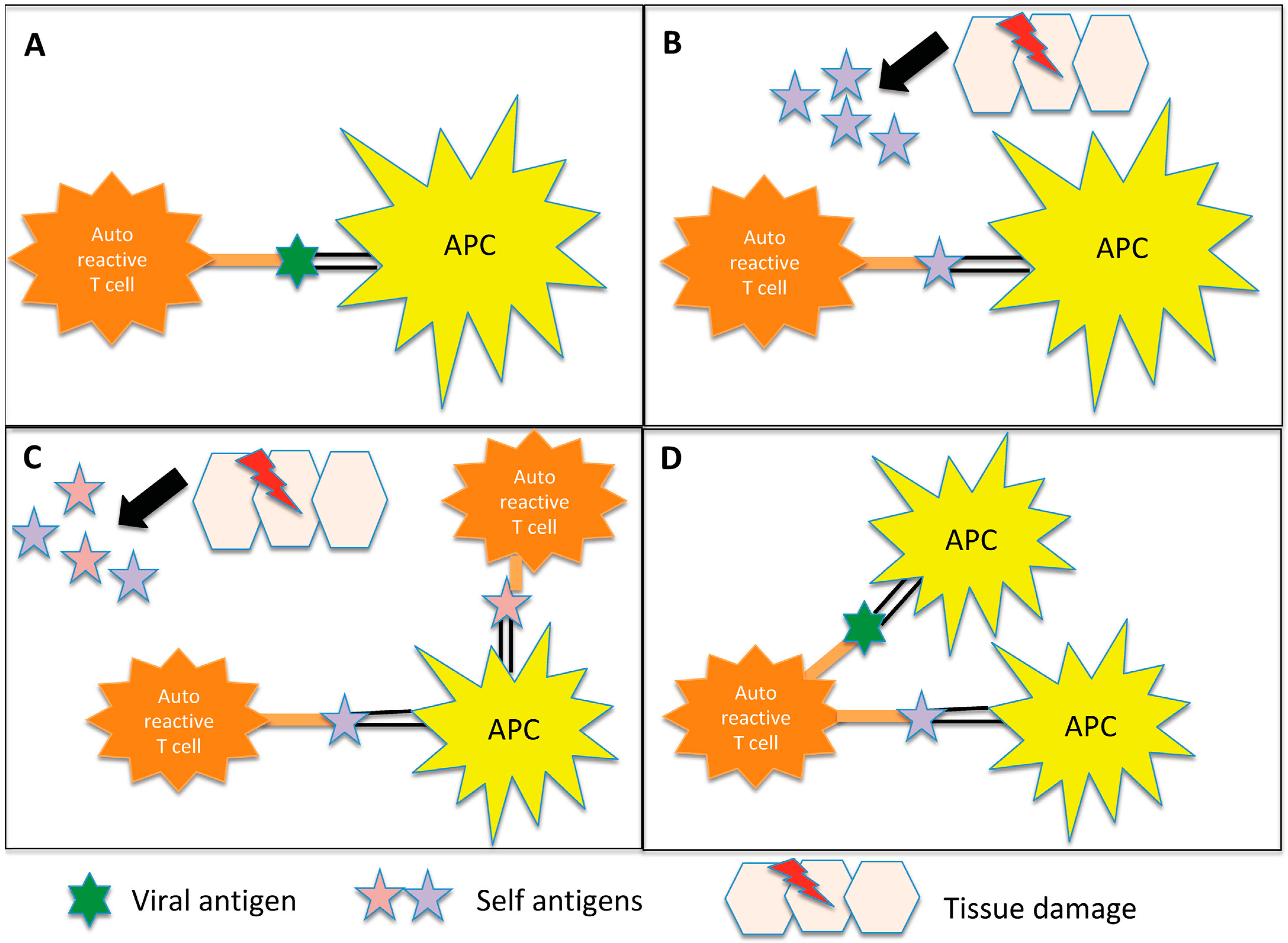{kind=link}
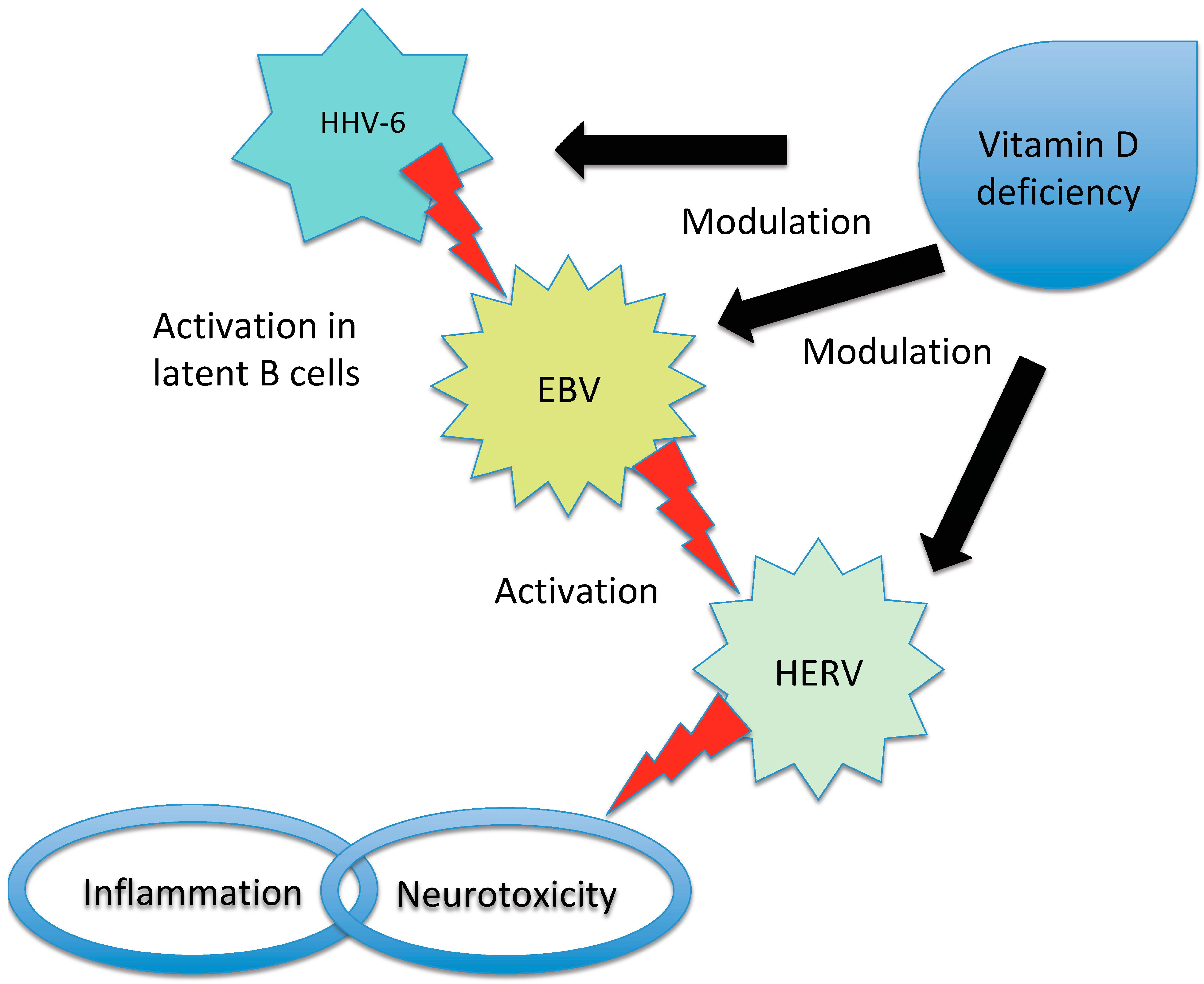{kind=link}
| MS | ||
|---|---|---|
| Infectious Agent | Evidence | References |
| EBV | Definitively established association between EBV infection and MS onset in a wide cohort study | [21] |
| High titers of anti-EBNA and VCA antibodies are observed in patients with MS | [25,26,27] | |
| EBV is a necessary causative agent in the pathogenesis of MS | [28] | |
| Serum titers of pre-onset anti-EBNA antibodies are strong markers of MS | [29] | |
| EBNA-1 recognized by MS patient sera induces signs of EAE in a murine model | [30] | |
| EBNA-1 peptides are cross recognized by anti-MBP antibodies | [31] | |
| Immunoreactivity against EBV proteins BRRF2 and EBNA-1 is higher in MS; OCBs belonging to MS patients bound both EBV proteins | [32] | |
| OCBs in CSF belonging to MS patients are able to bind EBNA-1 and EBNA-2 epitopes | [33] | |
| There is high-affinity molecular mimicry between EBNA-1 and GlialCAM in MS | [34] | |
| High titers of anti-EBNA increase the risk of MS and are observed between 15 and 20 years before the onset of the disease | [35] | |
| The risk of MS is notably increased after infectious mononucleosis | [36] | |
| There is evidence of EBV infection in brain-infiltrating B cells and plasma cells in MS | [37] | |
| Mutations in EBNA-2 could influence the host response to EBV | [38] | |
| HLA-DRB1*15:01 acts as coreceptor for EBV infection of B cells | [39] | |
| Specific EBNA-1 antibodies and HLA-DRB1*1501 interact in the MS risk | [40] | |
| EBNA-3 blocks the activation of vitamin D receptor-dependent genes | [41] | |
| EBNA-1 antibodies correlate with radiological disease activity | [42,43] | |
| The cellular immune response to EBV decreases during ocrelizumab treatment | [44] | |
| Teriflunomide inhibits cellular proliferation in EBV-transformed B cells | [45] | |
| HERV | The envelope protein of HERV-W has been detected in serum, brain, perivascular infiltrates and macrophages of patients with MS | [46,47,48] |
| HERV mRNA has been found in the brain lesions, CSF and blood cells of individuals with MS | [49,50] | |
| The expression of HERV is increased in patients with active MS | [51,52] | |
| There is evidence of molecular mimicry between HERV-W envelope protein and myelin proteins | [53] | |
| HERV may activate the host immune response by acting as an agonist of human toll-like receptor 4 | [54] | |
| The HERV-H envelope and gag proteins have been reported to be present in the serum of MS patients | [55] | |
| HERV-W could act as effector in MS pathogenesis through its activation during EBV infection | [56] | |
| EBV transactivates the HERV-K18 that encodes a superantigen | [57] | |
| HERV-W DNA copy number was found to be higher in MS patients and was inversely correlated with vitamin D level | [58] | |
| Interferon beta may decrease the expression of HERV-W | [59] | |
| Natalizumab inhibits the expression of HERV-W | [60] | |
| A new drug tested in a phase II clinical trial for MS, known as GNbAC1, is able to block the HERV-W-dependent inflammatory cascade | [61,62] | |
| HHV-6 | OCBs specific against HHV-6 have been identified in patients with MS | [63] |
| Pro-inflammatory cytokines are higher in HHV-6 infected patients and HHV-6 positivity is associated with higher disability | [64] | |
| Anti-HHV-6 IgG titers significantly predict subsequent relapse risk in MS | [65] | |
| The lymphoproliferative response to HHV-6A is increased in MS | [66] | |
| There is an increased prevalence of HHV-6A in MS | [67] | |
| MBP cross-reacts with HHV-6 antigens; thus, there is evidence of a molecular mimicry | [68] | |
| Increased serological response against HHV-6A is associated with the risk of MS | [69] | |
| There is an interaction between environmental factors and high titers of anti-HHV-6A antibodies in the risk of MS | [70] | |
| HHV-6A is a risk factor for MS | [71] | |
| Gut Microbiota | Gut bacteria from patients with MS have pro-inflammatory properties | [72] |
| Disease-modifying therapies alter gut microbial composition in MS | [73] | |
| Interferon beta can cause an increase in Prevotella | [74] | |
| Gut microbiota differs from MS and controls. Enterobacteriaceae and several Clostridium species are associated with progressive course and disability | [75] | |
| There is an alteration of gut microbiota in MS patients, with an over-representation of Saccharomyces and Aspergillus | [76] | |
| Fungi | First evidence of fungal infection in CNS tissue of MS patients, with detection of fungal DNA | [77] |
| The specific enzyme activity of Candida albicans is greater in MS patients and correlates with disease severity | [78] | |
| Fungal antigens and antibodies against several Candida species have been detected in CSF of MS patients | [79] | |
| Calprotectin levels in the CSF reflect disease activity | [80] | |
| Some improvement in MS symptoms was observed in MS patients after treatment with antifungal drugs | [81] | |
| MAP | MAP peptides are cross-recognized by anti-MBP antibodies | [31] |
| MAP is associated with MS in Sardinian population | [82,83] | |
| MAP is associated with MS in Japanese population | [84] | |
| Epitopes of MAP2694 homologous to TCR are highly recognized in MS | [85] | |
| Human IRF 5 homologous epitopes of MAP induce a specific immune response | [86] | |
| There is no association between the haplotypes predisposing to MS and MAP positivity | [87] | |
| CMV | CMV can intensify the symptoms in MS patients | [88] |
| CMV seropositivity is negatively associated with MS | [89,90] | |
| NMOSD | ||
|---|---|---|
| Infectious Agent | Evidence | References |
| TB | Case report: a patient with NMOSD experienced relapses following episodes of pulmonary TB | [155] |
| There is no association between NMO and pulmonary TB in a Chinese cohort | [156] | |
| There is an association between NMO and pulmonary TB in a South-African cohort | [157] | |
| Two cases of NMOSD with onset temporally related to pulmonary TB | [158] | |
| Antituberculosis treatment reduced relapses in patients with steroid-refractory NMO and led to neurological recovery | [159] | |
| Pulmonary TB was associated with NMOSD in 5 out of 24 Brazilian patients | [160] | |
| H. pylori | High titers of anti H. pylori antibodies are observed only in AQP4-positive patients | [161] |
| High titers of anti H. pylori antibodies are observed in both AQP4-positive and -negative patients, with stronger association with the first group | [162] | |
| A higher prevalence of H. pylori infection was described in AQP4-positive compared to AQP4-negative patients | [163] | |
| There is no difference between AQP4-negative patients and healthy controls in terms of the frequency of H. pylori infection | [164] | |
| EBV | Anti-EA IgG are higher in NMO patients than in MS patients and healthy controls, both in sera and CSF | [165] |
| NMO is associated with the reactivation of EBV | [166] | |
| HERV | Patients with NMOSD have lower levels of anti-HERV antibodies compared to patients with MS and to healthy individuals | [167,168] |
| Gut microbiota | Clostridium perfringens is abundant in individuals with NMO | [169] |
| There is a potential molecular mimicry between Clostridium perfrigens and AQP4 | [170] | |
| Fungi | High in vitro immune reactivity to Escherichia coli is correlated with disability in NMO | [148] |
| HIV | Characterization of six cases of patients with HIV developing NMOSD | [171] |
| HIV is able to activate several cellular lines of the immune system | [172] | |
| Characterization of two cases of patients with HIV developing NMO | [173] | |
| MOGAD | ||
|---|---|---|
| Infectious Agent | Evidence | References |
| HERV | Higher levels of antibodies against HER-W were present in MOGAD than in NMO | [168] |
| There is a cross-reactivity between MOG and HERV-W proteins | [193] | |
| Measles | Case report: ADEM associated with measles | [183] |
| Influenza | Case report: ADEM associated with influenza A H1N1 infection | [184] |
| Coronavirus | Case report: ADEM with detection of coronavirus in CNS | [186] |
| Enterovirus | Case report: ADEM with positive PCR for enterovirus in the CSF | [185] |
| Mycoplasma pneumoniae | Description of cases reporting MOGAD following Mycoplasma pneumoniae infection | [189,190] |
| Jamestown Canyon virus | Case report: MOGAD with bilateral corticospinal tract lesions following infection with Jamestown Canyon virus | [194] |
| Streptococcus | Case report: MOGAD transverse myelitis following brain abscess | [192] |
| EBV | Case report: anti-MOG positive ADEM following infectious mononucleosis | [187] |
| Herpes simplex | Case report: MOGAD (bilateral optic neuritis and meningoganglionitis) following a genital herpes simplex virus infection | [188] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frau, J.; Coghe, G.; Lorefice, L.; Fenu, G.; Cocco, E. The Role of Microorganisms in the Etiopathogenesis of Demyelinating Diseases. Life 2023, 13, 1309. https://doi.org/10.3390/life13061309
Frau J, Coghe G, Lorefice L, Fenu G, Cocco E. The Role of Microorganisms in the Etiopathogenesis of Demyelinating Diseases. Life. 2023; 13(6):1309. https://doi.org/10.3390/life13061309
Chicago/Turabian StyleFrau, Jessica, Giancarlo Coghe, Lorena Lorefice, Giuseppe Fenu, and Eleonora Cocco. 2023. "The Role of Microorganisms in the Etiopathogenesis of Demyelinating Diseases" Life 13, no. 6: 1309. https://doi.org/10.3390/life13061309
APA StyleFrau, J., Coghe, G., Lorefice, L., Fenu, G., & Cocco, E. (2023). The Role of Microorganisms in the Etiopathogenesis of Demyelinating Diseases. Life, 13(6), 1309. https://doi.org/10.3390/life13061309