

The Replicative DnaE Polymerase of Bacillus subtilis Recruits the Glycolytic Pyruvate Kinase (PykA) When Bound to Primed DNA Templates



Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning of the pykA Gene and Production and Purification of the Protein
2.1.1. Cloning of pykA
2.1.2. Expression of pykA
2.1.3. Purification of PykA
2.1.4. PykA Activity Assay
2.2. Cloning of a DNA Fragment Coding for the PEPut Domain of PykA and Production and Purification of the Peptide
2.2.1. Fragment Cloning
2.2.2. Production and Purification of PEPut
2.3. Cloning of DNA Fragments Coding for CAT or Mutated PykA, Expression, and Purification of the Proteins
2.4. Production and Purification of the B. subtilis DnaE Polymerase
2.5. Polymerase Assays
2.6. Surface Plasmon Resonance (SPR)
2.7. B. subtilis Strains and Growth Conditions
2.8. Microscopy
3. Results
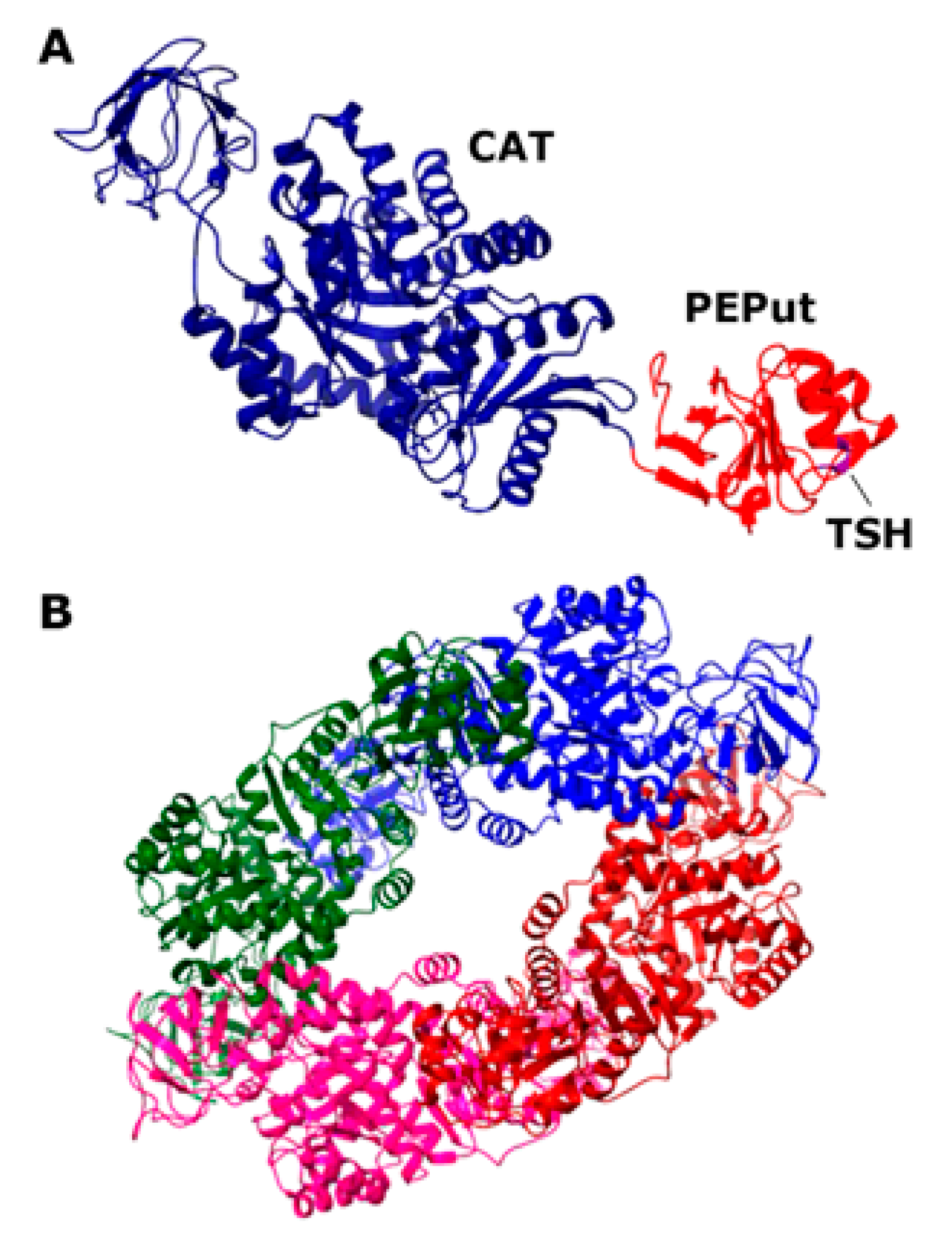
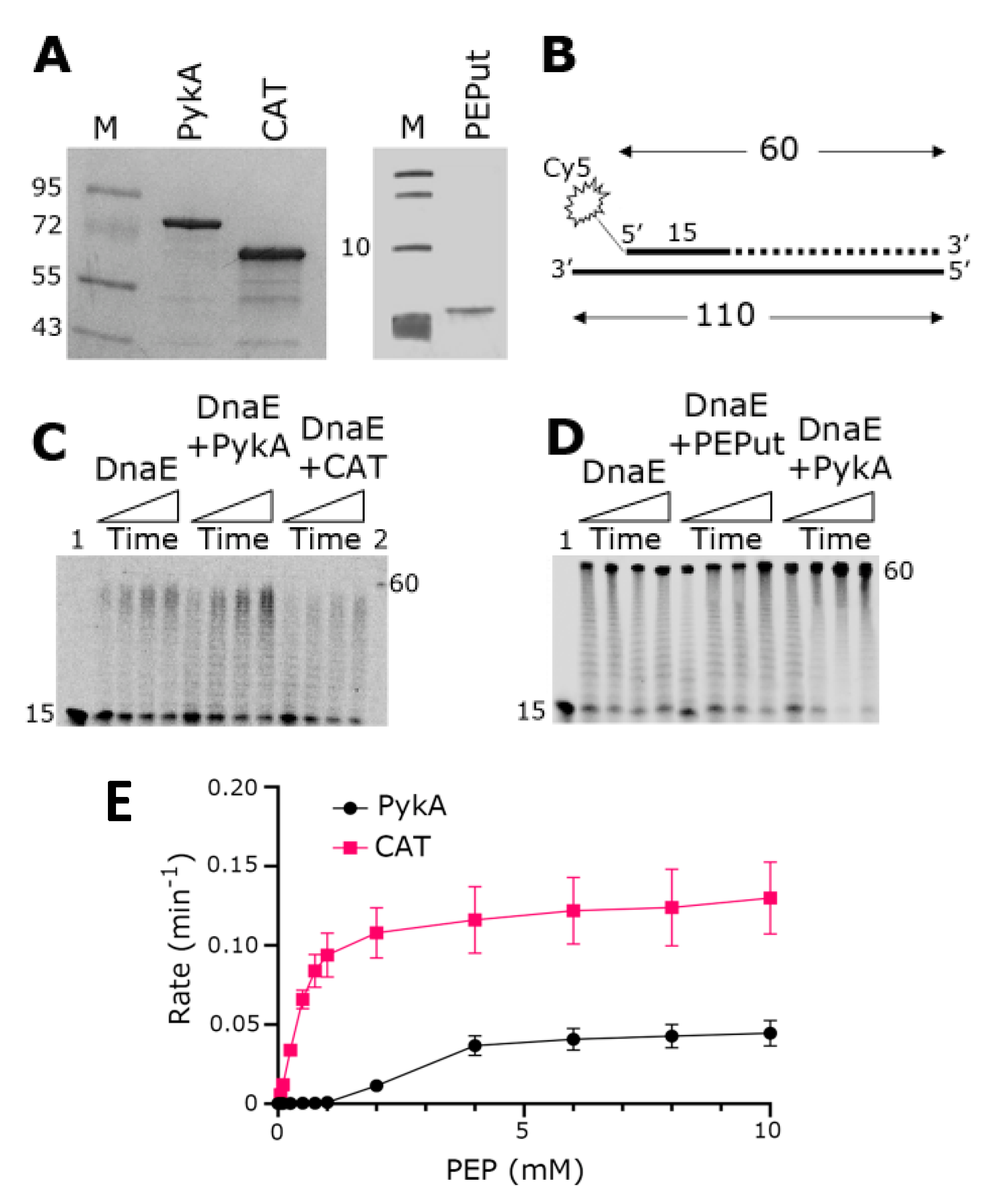
3.1. The CAT Domain Alone Elicits a Functional Effect on the DnaE Polymerase Activity and PEPut Modulates This Effect and PykA Catalytic Activity
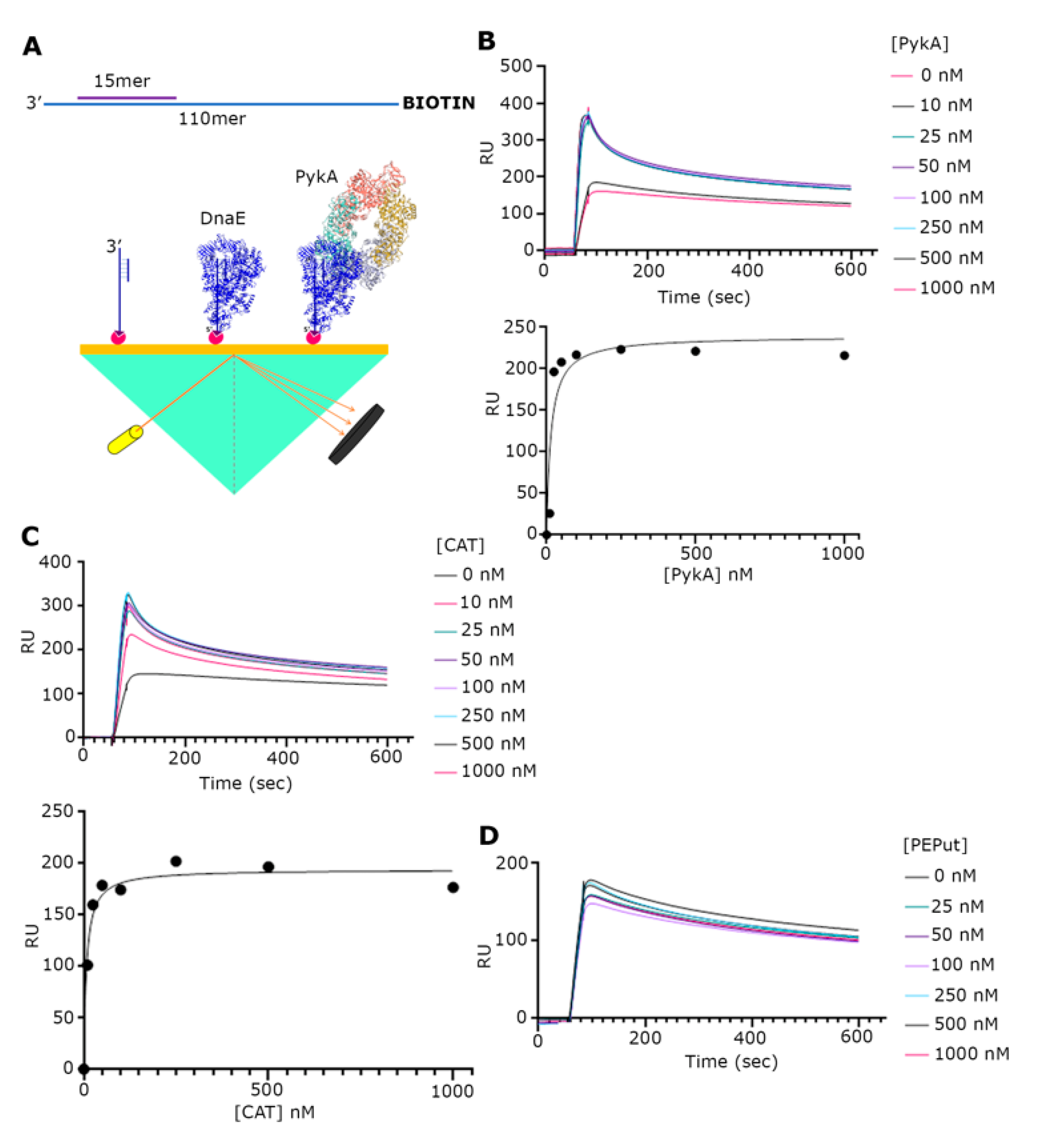
3.2. The CAT Domain Mediates the Physical Interaction with DnaE in a Complex with a Primed Template and PEPut Reduces the Strength of This Interaction
3.3. The TSH Motif of PEPut Impacts the Catalytic Activity of PykA but Not Its Effect on DnaE Activity
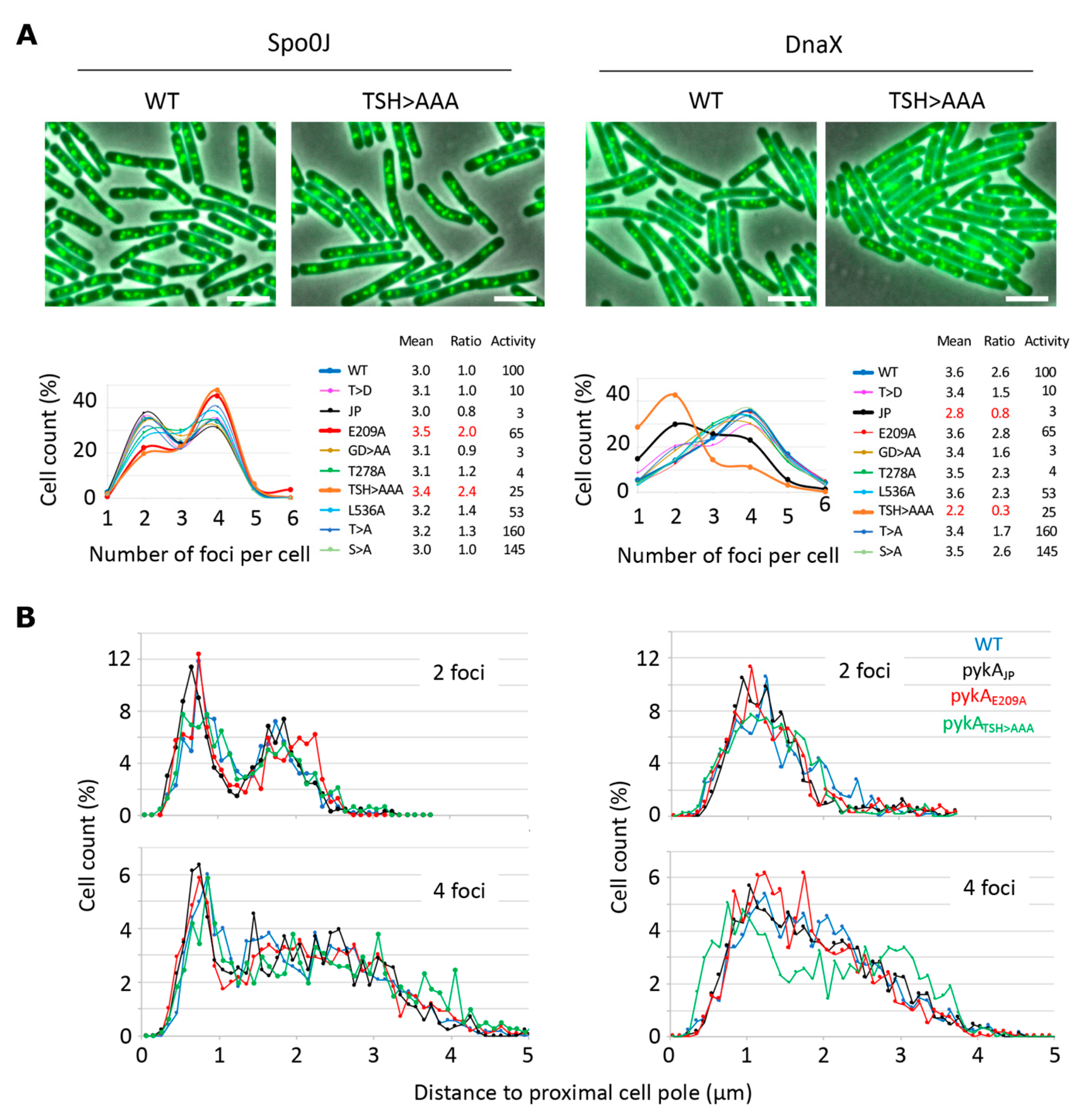
3.4. PEPut and Cat Mutations Impact Origin and Replisome Localization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, C.-L.; Bian, Y.-Y.; Xue, Y.; Liu, Z.-X.; Zhou, K.-Q.; Yao, C.-F.; Lin, Y.; Zou, H.-F.; Luo, F.-X.; Qu, Y.-Y.; et al. Pyruvate Kinase M2 Activates MTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016, 6, 21524. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Yu, D. Protein Kinase Function of Pyruvate Kinase M2 and Cancer. Cancer Cell Int. 2020, 20, 523. [Google Scholar] [CrossRef] [PubMed]
- Jannière, L.; Canceill, D.; Suski, C.; Kanga, S.; Dalmais, B.; Lestini, R.; Monnier, A.-F.; Chapuis, J.; Bolotin, A.; Titok, M.; et al. Genetic Evidence for a Link between Glycolysis and DNA Replication. PLoS ONE 2007, 2, e447. [Google Scholar] [CrossRef] [PubMed]
- Nouri, H.; Monnier, A.-F.; Fossum-Raunehaug, S.; Maciąg-Dorszyńska, M.; Cabin-Flaman, A.; Képès, F.; Węgrzyn, G.; Szalewska-Pałasz, A.; Norris, V.; Skarstad, K.; et al. Multiple Links Connect Central Carbon Metabolism to DNA Replication Initiation and Elongation in Bacillus subtilis. DNA Res. 2018, 25, 641–653. [Google Scholar] [CrossRef]
- Horemans, S.; Pitoulias, M.; Holland, A.; Pateau, E.; Lechaplais, C.; Ekaterina, D.; Perret, A.; Soultanas, P.; Janniere, L. Pyruvate Kinase, a Metabolic Sensor Powering Glycolysis, Drives the Metabolic Control of DNA Replication. BMC Biol. 2022, 20, 87. [Google Scholar] [CrossRef]
- Mathews, C.K. Deoxyribonucleotide Metabolism, Mutagenesis and Cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks: Error-prone repair of DNA DSBS. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef]
- Saxena, S.; Zou, L. Hallmarks of DNA Replication Stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Liu, X.; Gu, C.; Luo, R.; Chen, H.-F. Synergistic Allosteric Mechanism of Fructose-1,6-Bisphosphate and Serine for Pyruvate Kinase M2 via Dynamics Fluctuation Network Analysis. J. Chem. Inf. Model. 2016, 56, 1184–1192. [Google Scholar] [CrossRef]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine Is a Natural Ligand and Allosteric Activator of Pyruvate Kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef]
- Nakatsu, D.; Horiuchi, Y.; Kano, F.; Noguchi, Y.; Sugawara, T.; Takamoto, I.; Kubota, N.; Kadowaki, T.; Murata, M. L-Cysteine Reversibly Inhibits Glucose-Induced Biphasic Insulin Secretion and ATP Production by Inactivating PKM2. Proc. Natl. Acad. Sci. USA 2015, 112, E1067–E1076. [Google Scholar] [CrossRef]
- Schormann, N.; Hayden, K.L.; Lee, P.; Banerjee, S.; Chattopadhyay, D. An Overview of Structure, Function, and Regulation of Pyruvate Kinases. Protein Sci. 2019, 28, 1771–1784. [Google Scholar] [CrossRef]
- Sakai, H.; Suzuki, K.; Imahori, K. Purification and Properties of Pyruvate Kinase from Bacillus stearothermophilus. J. Biochem. 1986, 99, 1157–1167. [Google Scholar] [CrossRef]
- Morgan, H.P.; Zhong, W.; McNae, I.W.; Michels, P.A.M.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. Structures of Pyruvate Kinases Display Evolutionarily Divergent Allosteric Strategies. R. Soc. Open Sci. 2014, 1, 140120. [Google Scholar] [CrossRef]
- Sakai, H. Possible Structure and Function of the Extra C-Terminal Sequence of Pyruvate Kinase from Bacillus stearothermophilus. J. Biochem. 2004, 136, 471–476. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Saier, M.H. Phylogenetic Analysis of the Putative Phosphorylation Domain in the Pyruvate Kinase of Bacillus stearothermophilus. Res. Microbiol. 1995, 146, 713–719. [Google Scholar] [CrossRef]
- Alpert, C.A.; Frank, R.; Stueber, K.; Deutscher, J.; Hengstenberg, W. Phosphoenolpyruvate-Dependent Protein Kinase Enzyme I of Streptococcus faecalis: Purification and Properties of the Enzyme and Characterization of Its Active Center. Biochemistry 1985, 24, 959–964. [Google Scholar] [CrossRef]
- Teplyakov, A.; Lim, K.; Zhu, P.-P.; Kapadia, G.; Chen, C.C.H.; Schwartz, J.; Howard, A.; Reddy, P.T.; Peterkofsky, A.; Herzberg, O. Structure of Phosphorylated Enzyme I, the Phosphoenolpyruvate:Sugar Phosphotransferase System Sugar Translocation Signal Protein. Proc. Natl. Acad. Sci. USA 2006, 103, 16218–16223. [Google Scholar] [CrossRef]
- Goss, N.H.; Evans, C.T.; Wood, H.G. Pyruvate Phosphate Dikinase: Sequence of the Histidyl Peptide, the Pyrophosphoryl and Phosphoryl Carrier. Biochemistry 1980, 19, 5805–5809. [Google Scholar] [CrossRef]
- Herzberg, O.; Chen, C.C.; Kapadia, G.; McGuire, M.; Carroll, L.J.; Noh, S.J.; Dunaway-Mariano, D. Swiveling-Domain Mechanism for Enzymatic Phosphotransfer between Remote Reaction Sites. Proc. Natl. Acad. Sci. USA 1996, 93, 2652–2657. [Google Scholar] [CrossRef] [PubMed]
- Tolentino, R.; Chastain, C.; Burnell, J. Identification of the Amino Acid Involved in the Regulation of Bacterial Pyruvate, Orthophosphate Dikinase and Phosphoenolpyruvate Synthetase. Adv. Biol. Chem. 2013, 3, 12–21. [Google Scholar] [CrossRef]
- Burnell, J.N.; Chastain, C.J. Cloning and Expression of Maize-Leaf Pyruvate, Pi Dikinase Regulatory Protein Gene. Biochem. Biophys. Res. Commun. 2006, 345, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Burnell, J.N. Cloning and Characterization of Escherichia Coli DUF299: A Bifunctional ADP-Dependent Kinase—Pi-Dependent Pyrophosphorylase from Bacteria. BMC Biochem. 2010, 11, 1. [Google Scholar] [CrossRef]
- Burnell, J.N.; Hatch, M.D. Regulation of C4 Photosynthesis: Identification of a Catalytically Important Histidine Residue and Its Role in the Regulation of Pyruvate,Pi Dikinase. Arch. Biochem. Biophys. 1984, 231, 175–182. [Google Scholar] [CrossRef]
- Eymann, C.; Dreisbach, A.; Albrecht, D.; Bernhardt, J.; Becher, D.; Gentner, S.; Tam, L.T.; Büttner, K.; Buurman, G.; Scharf, C.; et al. A Comprehensive Proteome Map of Growing Bacillus subtilis Cells. Proteomics 2004, 4, 2849–2876. [Google Scholar] [CrossRef]
- Mader, U.; Schmeisky, A.G.; Florez, L.A.; Stulke, J. SubtiWiki—A Comprehensive Community Resource for the Model Organism Bacillus subtilis. Nucleic Acids Res. 2012, 40, D1278–D1287. [Google Scholar] [CrossRef]
- Reiland, S.; Messerli, G.; Baerenfaller, K.; Gerrits, B.; Endler, A.; Grossmann, J.; Gruissem, W.; Baginsky, S. Large-Scale Arabidopsis Phosphoproteome Profiling Reveals Novel Chloroplast Kinase Substrates and Phosphorylation Networks. Plant Physiol. 2009, 150, 889–903. [Google Scholar] [CrossRef]
- Suzuki, K.; Ito, S.; Shimizu-Ibuka, A.; Sakai, H. Crystal Structure of Pyruvate Kinase from Geobacillus stearothermophilus. J. Biochem. 2008, 144, 305–312. [Google Scholar] [CrossRef]
- Rannou, O.; Le Chatelier, E.; Larson, M.A.; Nouri, H.; Dalmais, B.; Laughton, C.; Jannière, L.; Soultanas, P. Functional Interplay of DnaE Polymerase, DnaG Primase and DnaC Helicase within a Ternary Complex, and Primase to Polymerase Hand-off during Lagging Strand DNA Replication in Bacillus subtilis. Nucleic Acids Res. 2013, 41, 5303–5320. [Google Scholar] [CrossRef]
- Paschalis, V.; Le Chatelier, E.; Green, M.; Képès, F.; Soultanas, P.; Janniere, L. Interactions of the Bacillus subtilis DnaE Polymerase with Replisomal Proteins Modulate Its Activity and Fidelity. Open Biol. 2017, 7, 170146. [Google Scholar] [CrossRef]
- Simmons, L.A.; Grossman, A.D.; Walker, G.C. Replication Is Required for the RecA Localization Response to DNA Damage in Bacillus Subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 1360–1365. [Google Scholar] [CrossRef]
- Monahan, L.G.; Hajduk, I.V.; Blaber, S.P.; Charles, I.G.; Harry, E.J. Coordinating Bacterial Cell Division with Nutrient Availability: A Role for Glycolysis. mBio 2014, 5, e00935-14. [Google Scholar] [CrossRef]
- Weart, R.B.; Lee, A.H.; Chien, A.-C.; Haeusser, D.P.; Hill, N.S.; Levin, P.A. A Metabolic Sensor Governing Cell Size in Bacteria. Cell 2007, 130, 335–347. [Google Scholar] [CrossRef]
- Sanders, G.M.; Dallmann, H.G.; McHenry, C.S. Reconstitution of the B. subtilis Replisome with 13 Proteins Including Two Distinct Replicases. Mol. Cell 2010, 37, 273–281. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Matthews, L.A.; Simmons, L.A.; Biteen, J.S. Dynamic Exchange of Two Essential DNA Polymerases during Replication and after Fork Arrest. Biophys. J. 2019, 116, 684–693. [Google Scholar] [CrossRef]
- Hernández-Tamayo, R.; Oviedo-Bocanegra, L.M.; Fritz, G.; Graumann, P.L. Symmetric Activity of DNA Polymerases at and Recruitment of Exonuclease ExoR and of PolA to the Bacillus subtilis Replication Forks. Nucleic Acids Res. 2019, 47, 8521–8536. [Google Scholar] [CrossRef]
- Murray, H.; Koh, A. Multiple Regulatory Systems Coordinate DNA Replication with Cell Growth in Bacillus subtilis. PLoS Genet. 2014, 10, e1004731. [Google Scholar] [CrossRef]
- Maciąg, M.; Nowicki, D.; Janniere, L.; Szalewska-Pałasz, A.; Węgrzyn, G. Genetic Response to Metabolic Fluctuations: Correlation between Central Carbon Metabolism and DNA Replication in Escherichia coli. Microb. Cell Factories 2011, 10, 19. [Google Scholar] [CrossRef]
- Maciąg-Dorszyńska, M.; Ignatowska, M.; Jannière, L.; Węgrzyn, G.; Szalewska-Pałasz, A. Mutations in Central Carbon Metabolism Genes Suppress Defects in Nucleoid Position and Cell Division of Replication Mutants in Escherichia coli. Gene 2012, 503, 31–35. [Google Scholar] [CrossRef]
- Krause, K.; Maciąg-Dorszyńska, M.; Wosinski, A.; Gaffke, L.; Morcinek-Orłowska, J.; Rintz, E.; Bielańska, P.; Szalewska-Pałasz, A.; Muskhelishvili, G.; Węgrzyn, G. The Role of Metabolites in the Link between DNA Replication and Central Carbon Metabolism in Escherichia Coli. Genes 2020, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Bergé, M.; Pezzatti, J.; González-Ruiz, V.; Degeorges, L.; Mottet-Osman, G.; Rudaz, S.; Viollier, P.H. Bacterial Cell Cycle Control by Citrate Synthase Independent of Enzymatic Activity. eLife 2020, 9, e52272. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.; Landoulsi, A.; Kohiyama, M. A Novel Role for CAMP in the Control of the Activity of the E. coli Chromosome Replication Initiator Protein, DnaA. Cell 1988, 55, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhou, A.; Li, S.; Ni, J.; Tao, J.; Lu, J.; Wan, B.; Li, S.; Zhang, J.; Zhao, S.; et al. Reversible Lysine Acetylation Is Involved in DNA Replication Initiation by Regulating Activities of Initiator DnaA in Escherichia coli. Sci. Rep. 2016, 6, 30837. [Google Scholar] [CrossRef]
- Vogelauer, M.; Rubbi, L.; Lucas, I.; Brewer, B.J.; Grunstein, M. Histone Acetylation Regulates the Time of Replication Origin Firing. Mol. Cell 2002, 10, 1223–1233. [Google Scholar] [CrossRef]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Zheng, L.; Roeder, R.G.; Luo, Y. S Phase Activation of the Histone H2B Promoter by OCA-S, a Coactivator Complex That Contains GAPDH as a Key Component. Cell 2003, 114, 255–266. [Google Scholar] [CrossRef]
- Dai, R.-P.; Yu, F.-X.; Goh, S.-R.; Chng, H.-W.; Tan, Y.-L.; Fu, J.-L.; Zheng, L.; Luo, Y. Histone 2B (H2B) Expression Is Confined to a Proper NAD+/NADH Redox Status. J. Biol. Chem. 2008, 283, 26894–26901. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Jiang, H.; Xia, Y.; Zheng, Y.; Li, J.; Huang, B.-J.; Fang, J.; Qian, C.-N.; Jiang, T.; et al. Nuclear PGK1 Alleviates ADP-Dependent Inhibition of CDC7 to Promote DNA Replication. Mol. Cell 2018, 72, 650–660.e8. [Google Scholar] [CrossRef]
- Konieczna, A.; Szczepańska, A.; Sawiuk, K.; Węgrzyn, G.; Łyżeń, R. Effects of Partial Silencing of Genes Coding for Enzymes Involved in Glycolysis and Tricarboxylic Acid Cycle on the Enterance of Human Fibroblasts to the S Phase. BMC Cell Biol. 2015, 16, 16. [Google Scholar] [CrossRef]
- Fornalewicz, K.; Wieczorek, A.; Węgrzyn, G.; Łyżeń, R. Silencing of the Pentose Phosphate Pathway Genes Influences DNA Replication in Human Fibroblasts. Gene 2017, 635, 33–38. [Google Scholar] [CrossRef]
- Wieczorek, A.; Fornalewicz, K.; Mocarski, Ł.; Łyżeń, R.; Węgrzyn, G. Double Silencing of Relevant Genes Suggests the Existence of the Direct Link between DNA Replication/Repair and Central Carbon Metabolism in Human Fibroblasts. Gene 2018, 650, 1–6. [Google Scholar] [CrossRef]
- Grosse, F.; Nasheuer, H.-P.; Scholtissek, S.; Schomburg, U. Lactate Dehydrogenase and Glyceraldehyde-Phosphate Dehydrogenase Are Single-Stranded DNA-Binding Proteins That Affect the DNA-Polymerase-Alpha-Primase Complex. Eur. J. Biochem. 1986, 160, 459–467. [Google Scholar] [CrossRef]
- Popanda, O.; Fox, G.; Thielmann, H.W. Modulation of DNA Polymerases α, δ and ε by Lactate Dehydrogenase and 3-Phosphoglycerate Kinase. Biochim. Biophys. Acta BBA—Gene Struct. Expr. 1998, 1397, 102–117. [Google Scholar] [CrossRef]
- Jindal, H.K.; Vishwanatha, J.K. Functional Identity of a Primer Recognition Protein as Phosphoglycerate Kinase. J. Biol. Chem. 1990, 265, 6540–6543. [Google Scholar] [CrossRef]
- Schaechter, M.; MaalOe, O.; Kjeldgaard, N.O. Dependency on Medium and Temperature of Cell Size and Chemical Composition during Balanced Growth of Salmonella typhimurium. J. Gen. Microbiol. 1958, 19, 592–606. [Google Scholar] [CrossRef]
- Stokke, C.; Flåtten, I.; Skarstad, K. An Easy-To-Use Simulation Program Demonstrates Variations in Bacterial Cell Cycle Parameters Depending on Medium and Temperature. PLoS ONE 2012, 7, e30981. [Google Scholar] [CrossRef]
- Helmstetter, C.E. Timing of Synthetic Activities in the Cell Cycle. In Escherichia coli and Salmonella: Cellular and Molecular Biology; ASM Press: Washington, DC, USA, 1996; pp. 1627–1639. [Google Scholar]
- Sharpe, M.E.; Hauser, P.M.; Sharpe, R.G.; Errington, J. Bacillus subtilis Cell Cycle as Studied by Fluorescence Microscopy: Constancy of Cell Length at Initiation of DNA Replication and Evidence for Active Nucleoid Partitioning. J. Bacteriol. 1998, 180, 547–555. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PykA R32A Fwd | GAACGTGGCTgcATTAAACTTTTC |
| PykA R32A Rev | ATTCCTGACTCCATTAATTTC |
| PykA G245A D246A Fwd | GGTTGCACGCgcagcaTTAGGTGTGG |
| PykA G245A D246A Rev | ATTAAGCCGTCAGACACTTC |
| PykA TSH T537D Fwd | AGGCGGTTTGgatAGCCATGCTG |
| PykA TSH T537D Rev | TCTTCTGTAATAAGAGCAGAC |
| PykA TSH S538D Fwd | CGGTTTGACTgatCATGCTGCGG |
| PykA TSH S538D Rev | CCTTCTTCTGTAATAAGAGC |
| PykA TSH H539D Fwd | TTTGACTAGCgatGCTGCGGTAG |
| PykA TSH H539D Rev | CCGCCTTCTTCTGTAATAAG |
| FWD PykA_TSH DDD | CGGTTTGGATgatgATGCTGCGGTAG |
| REV PykA_TSH DDD | CCTTCTTCTGTAATAAGAGC |
| PykA CAT fwd | TAATAACATTGGAAGTGGATAAC |
| PykA CAT rev | GCCGACAGTATGAACCTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holland, A.; Pitoulias, M.; Soultanas, P.; Janniere, L. The Replicative DnaE Polymerase of Bacillus subtilis Recruits the Glycolytic Pyruvate Kinase (PykA) When Bound to Primed DNA Templates. Life 2023, 13, 965. https://doi.org/10.3390/life13040965
Holland A, Pitoulias M, Soultanas P, Janniere L. The Replicative DnaE Polymerase of Bacillus subtilis Recruits the Glycolytic Pyruvate Kinase (PykA) When Bound to Primed DNA Templates. Life. 2023; 13(4):965. https://doi.org/10.3390/life13040965
Chicago/Turabian StyleHolland, Alexandria, Matthaios Pitoulias, Panos Soultanas, and Laurent Janniere. 2023. "The Replicative DnaE Polymerase of Bacillus subtilis Recruits the Glycolytic Pyruvate Kinase (PykA) When Bound to Primed DNA Templates" Life 13, no. 4: 965. https://doi.org/10.3390/life13040965
APA StyleHolland, A., Pitoulias, M., Soultanas, P., & Janniere, L. (2023). The Replicative DnaE Polymerase of Bacillus subtilis Recruits the Glycolytic Pyruvate Kinase (PykA) When Bound to Primed DNA Templates. Life, 13(4), 965. https://doi.org/10.3390/life13040965