
Aiding Cancer’s “Sweet Tooth”: Role of Hexokinases in Metabolic Reprogramming

 ,
, 
Abstract
1. Introduction
1.1. General Characteristics and Distribution
1.2. Regulation of Hexokinase Expression
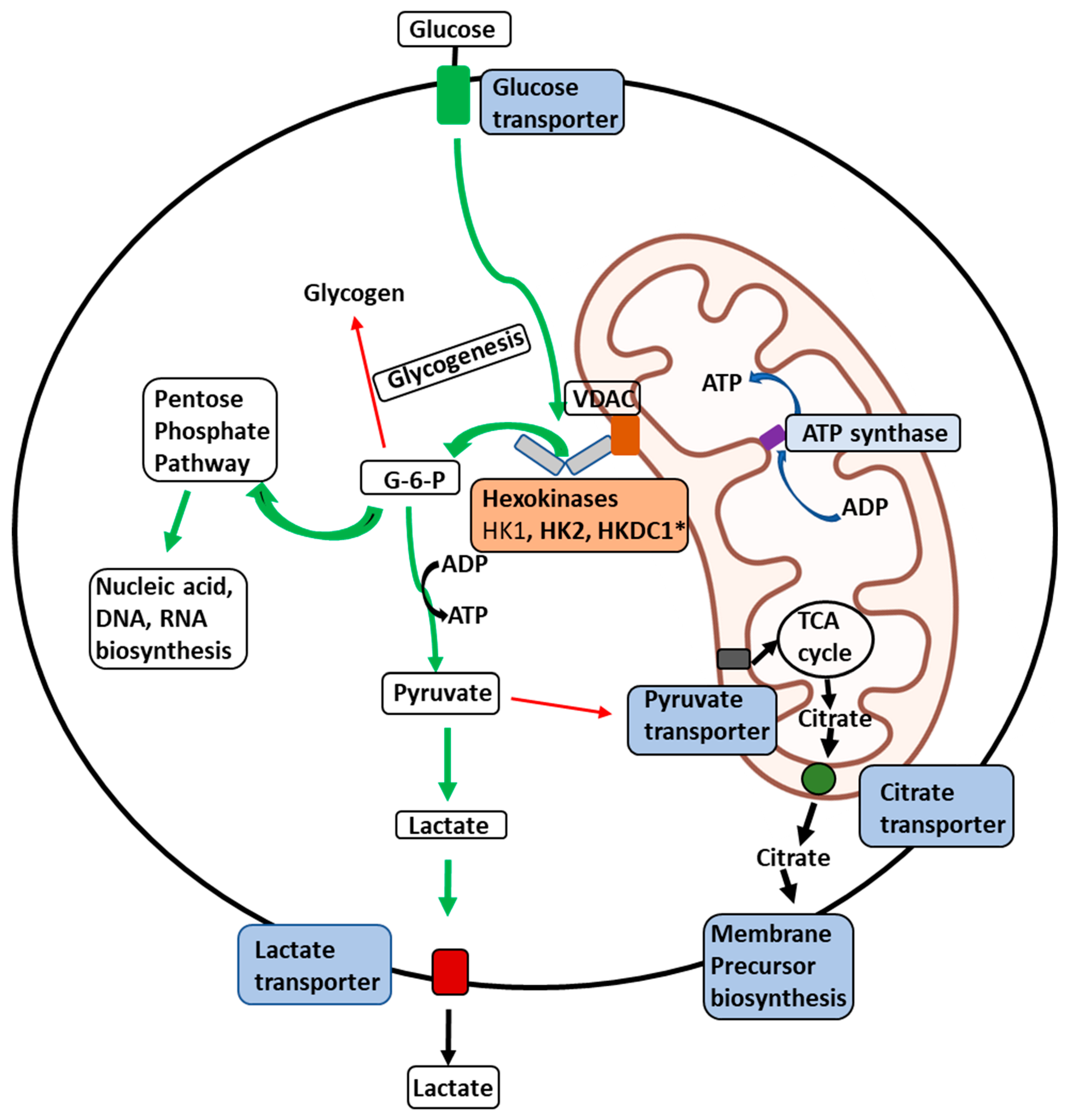
1.3. Regulation of Hexokinase Activity
1.4. Differences in Subcellular Localization
1.5. Roles of Hexokinases in Cancer-Mediated Metabolic Reprogramming
2. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Katzen, H.M.; Schimke, R.T. Multiple forms of hexokinase in the rat: Tissue distribution, age dependency, and properties. Proc. Natl. Acad. Sci. USA 1965, 54, 1218–1225. [Google Scholar] [CrossRef]
- Sebastian, S.; Hoebee, B.; Hande, M.; Kenkare, U.; Natarajan, A. Assignment of hexokinase types 1, 2, 3 (Hk1, 2, 3) and glucokinase (Gck) to rat chromosome band 20q11, 4q34, 17q12 and 14q21 respectively, by in situ hybridization. Cytogenet. Cell Genet. 1997, 77, 266–267. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206 Pt 13, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Matschinsky, F.M.; Wilson, D.F. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef]
- Wyatt, E.; Wu, R.; Rabeh, W.; Park, H.W.; Ghanefar, M.; Ardehali, H. Regulation and cytoprotective role of hexokinase III. PLoS ONE 2010, 5, e13823. [Google Scholar] [CrossRef]
- Cárdenas, M.L.; Cornish-Bowden, A.; Ureta, T. Evolution and regulatory role of the hexokinases. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1401, 242–264. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Magnuson, M.A. Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog. Hormone Res. 2001, 56, 195–217. [Google Scholar] [CrossRef]
- Ludvik, A.E.; Pusec, C.M.; Priyadarshini, M.; Angueira, A.R.; Guo, C.; Lo, A.; Hershen-house, K.S.; Yang, G.Y.; Ding, X.; Reddy, T.E.; et al. Is a novel hexokinase involved in whole-body glucose use. Endocrinology 2016, 157, 3452–3461. [Google Scholar] [CrossRef]
- Pusec, C.M.; De Jesus, A.; Khan, M.W.; Terry, A.R.; Ludvik, A.E.; Xu, K.; Giancola, N.; Pervaiz, H.; Smith, E.D.; Ding, X.; et al. Hepatic HKDC1 expression contributes to liver metabolism. Endocrinology 2019, 160, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.W.; Priyadarshini, M.; Cordoba-Chacon, J.; Becker, T.C.; Layden, B.T. Hepatic hexokinase domain containing 1 (HKDC1) improves whole body glucose tolerance and insulin sensitivity in pregnant mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 678–687. [Google Scholar] [CrossRef]
- Khan, M.W.; Terry, A.R.; Priyadarshini, M.; Ilievski, V.; Farooq, Z.; Guzman, G.; Cordoba-Chacon, J.; Ben-Sahra, I.; Wicksteed, B.; Layden, B.T. The hexokinase “HKDC1” interaction with the mitochondria is essential for liver cancer progression. Cell Death Dis. 2022, 28, 660. [Google Scholar] [CrossRef]
- Khan, M.W.; Ding, X.; Cotler, S.J.; Clarke, M.; Layden, B.T. Studies on the tissue localization of HKDC1, a putative novel fifth hexokinase, in humans. J. Histochem. Cytochem. 2018, 66, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zapater, J.L.; Lednovich, K.R.; Khan, M.W.; Pusec, C.M.; Layden, B.T. Hexokinase domain-containing protein-1 in metabolic diseases and beyond. Trends Endocrinol. Metab. 2022, 33, 72–84. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Hivert, M.F.; Armstrong, L.L.; Morrison, J.; Guo, C.; Lowe, L.P.; Scheftner, D.A.; Pluzhnikov, A.; Levine, D.M.; et al. Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies. Diabetes 2013, 62, 3282–3291. [Google Scholar] [CrossRef]
- Irwin, D.M.; Tan, H. Molecular evolution of the vertebrate hexokinase gene family: Identification of a conserved fifth vertebrate hexokinase gene. Comp. Biochem. Physiol. Part D Genomics Proteomics 2008, 3, 96–107. [Google Scholar] [CrossRef]
- Guo, C.; Ludvik, A.E.; Arlotto, M.E.; Hayes, M.G.; Armstrong, L.L.; Scholtens, D.M. Coordinated regulatory variation associated with gestational hyperglycemia regulates expression of the novel hexokinase HKDC1. Nat. Commun. 2015, 6, 6069. [Google Scholar] [CrossRef] [PubMed]
- Colowick, S.P. The hexokinases. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1973; Volume 9, pp. 1–48. [Google Scholar]
- Easterby, J.S.; O’Brien, M.J. Purification and properties of pig-heart hexokinase. Eur. J. Biochem. 1973, 38, 201–211. [Google Scholar] [CrossRef] [PubMed]
- White, T.K.; Wilson, J.E. Isolation and characterization of the discrete N- and C-terminal halves of rat brain hexokinase: Retention of full catalytic activity in the isolated C-terminal half. Arch. Biochem. Biophys. 1989, 274, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.K.; Filburn, C.R.; Pedersen, P.L. Structure/function relationships in hexokinase. Site-directed mutational analyses and characterization of overexpressed fragments implicate different functions for the N- and C-terminal halves of the enzyme. J. Biol. Chem. 1993, 268, 18259–18266. [Google Scholar] [CrossRef]
- Ureta, T.; Medina, C.; Preller, A. The evolution of hexokinases. Arch. Biol. Med. Exp. 1987, 20, 343–357. [Google Scholar] [PubMed]
- Tsai, H.J. Functional organization and evolution of mammalian hexokinases: Mutations that caused the loss of catalytic activity in N-terminal halves of type I and type III isozymes. Arch. Biochem. Biophys. 1999, 369, 149–156. [Google Scholar] [CrossRef]
- Kawai, S. Hypothesis: Structures, evolution, and ancestor of glucose kinases in the hexokinase family. J. Biosci. Bioeng. 2005, 99, 320–330. [Google Scholar] [CrossRef]
- Heikkinen, S.; Suppola, S.; Malkki, M.; Deeb, S.S. Mouse hexokinase II gene: Structure, cDNA, promoter analysis, and expression pattern. Mamm. Genome 2000, 11, 91–96. [Google Scholar] [CrossRef]
- Kwee, S.A.; Hernandez, B.; Chan, O.; Wong, L. Choline kinase alpha and hexokinase-2 protein expression in hepatocellular carcinoma: Association with survival. PLoS ONE 2012, 7, e46591. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Li, Y.; Tang, Z.; Kong, W. Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumor Biol. 2014, 35, 3743–3753. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhang, Y.; Wang, J.; Deng, Y.; Lin, D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 2016, 16, 38. [Google Scholar] [CrossRef]
- Botzer, L.E.; Maman, S.; Sagi-Assif, O.; Meshel, T.; Nevo, I.; Yron, I.; Witz, I.P. Hexokinase 2 is a determinant of neuroblastoma metastasis. Br. J. Cancer 2016, 114, 759–766. [Google Scholar] [CrossRef]
- Ogawa, H.; Nagano, H.; Konno, M.; Eguchi, H.; Koseki, J.; Kawamoto, K.; Nishida, N.; Colvin, H.; Tomokuni, A.; Tomimaru, Y.; et al. The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in patients with pancreatic cancer. Mol. Clin. Oncol. 2015, 3, 563–571. [Google Scholar] [CrossRef]
- He, H.C.; Bi, X.C.; Zheng, Z.W.; Dai, Q.S.; Han, Z.D.; Liang, Y.X.; Ye, Y.K.; Zeng, G.H.; Zhu, G.; Zhong, W.D. Real-time quantitative RT-PCR assessment of PIM-1 and hK2 mRNA expression in benign prostate hyperplasia and prostate cancer. Med. Oncol. 2009, 26, 303–308. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Giannoni, E.; Fearns, A.; Ribas, R.; Gao, Q.; Taddei, M.L.; Pintus, G.; Dowsett, M.; Isacke, C.M.; Martin, L.A. miR-155 drives metabolic reprogramming of ER+ breast cancer cells following long-term estrogen deprivation and predicts clinical response to aromatase inhibitors. Cancer Res. 2016, 76, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- van ‘t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Liu, X.; Miao, W.; Huang, M.; Li, L.; Dai, X.; Wang, Y. Elevated hexokinase II expression confers acquired resistance to 4-hydroxytamoxifen in breast cancer cells. Mol. Cell Proteomics 2019, 18, 2273–2284. [Google Scholar] [CrossRef]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R.; et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009, 7, 1438–1445. [Google Scholar] [CrossRef]
- Federzoni, E.A.; Humbert, M.; Torbett, B.E.; Behre, G.; Fey, M.F.; Tschan, M.P. CEBPA-dependent HK3 and KLF5 expression in primary AML and during AML differentiation. Sci. Rep. 2014, 4, 4261. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.M. Glucokinase, glucose homeostasis, and diabetes mellitus. Curr. Diab. Rep. 2005, 5, 171–176. [Google Scholar] [CrossRef]
- Iynedjian, P.B. Mammalian glucokinase and its gene. Biochem. J. 1993, 293 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Ferre, T.; Riu, E.; Bosch, F.; Valera, A. Evidence from transgenic mice that glucokinase is rate limiting for glucose utilization in the liver. FASEB J. 1996, 10, 1213–1218. [Google Scholar] [CrossRef]
- Wilson, J.E. Hexokinases. Rev. Physiol. Biochem. Pharmacol. 1995, 126, 65–198. [Google Scholar] [CrossRef] [PubMed]
- Matschinsky, F.M. Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes 1990, 39, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Velho, G.; Froguel, P.; Clement, K.; Pueyo, M.E.; Rakotoambinina, B.; Zouali, H.; Passa, P.; Cohen, D.; Robert, J.J. Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet 1992, 340, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.M.; Sturis, J.; Clement, K.; Vionnet, N.; Pueyo, M.E.; Stoffel, M.; Takeda, J.; Passa, P.; Cohen, D.; Bell, G.I.; et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J. Clin. Investig. 1994, 93, 1120–1130. [Google Scholar] [CrossRef]
- Velho, G.; Petersen, K.F.; Perseghin, G.; Hwang, J.H.; Rothman, D.L.; Pueyo, M.E.; Cline, G.W.; Froguel, P.; Shulman, G.I. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J. Clin. Investig. 1996, 98, 1755–1761. [Google Scholar] [CrossRef]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Walensky, L.D.; Zhang, C.Y.; Choi, C.S.; Fisher, J.K.; Molina, A.J.; Datta, S.R.; Pitter, K.L.; Bird, G.H.; Wikstrom, J.D.; et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 2008, 14, 144–153. [Google Scholar] [CrossRef]
- Chen, X.; Lv, Y.; Sun, Y.; Zhang, H.; Xie, W.; Zhong, L.; Chen, Q.; Li, M.; Li, L.; Feng, J.; et al. PGC1β regulates breast tumor growth and metastasis by SREBP1-mediated HKDC1 expression. Front. Oncol. 2019, 9, 290. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, J.; Wu, J.; Yu, Z.; Zhang, W.; Chen, Y.; Yao, P.; Zhang, H. HKDC1 C-terminal based peptides inhibit extranodal natural killer/T-cell lymphoma by modulation of mitochondrial function and EBV suppression. Leukemia 2020, 34, 2736–2748. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.J.; Kim, J.; Yun, M.; Park, J.H.; Lee, J.D. Enzymatic properties of the N- and C-terminal halves of human hexokinase, II. BMB Rep. 2009, 42, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L.; Tuszynski, J.A. Quantum metabolism explains the allometric scaling of metabolic rates. J. R. Soc. Interface 2010, 7, 507–514. [Google Scholar] [CrossRef]
- Printz, R.L.; Koch, S.; Potter, L.R.; O’Doherty, R.M.; Tiesinga, J.J.; Moritz, S.; Granner, D.K. Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J. Biol. Chem. 1993, 268, 5209–5219. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Liu, W.; Wilson, J.E. Isolation of the promoter for Type I hexokinase from rat. Arch. Biochem. Biophys. 1996, 335, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wilson, J.E. Two Sp sites are important cis elements regulating the upstream promoter region of the gene for rat Type I hexokinase. Arch. Biochem. Biophys. 1997, 346, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells. Isolation, sequence, and activity of the promoter for Type II hexokinase. J. Biol. Chem. 1995, 270, 16918–16925. [Google Scholar] [CrossRef] [PubMed]
- Osawa, H.; Robey, R.B.; Printz, R.L.; Granner, D.K. Identification and characterization of basal and cyclic AMP response elements in the promoter of the rat hexokinase II gene. J. Biol. Chem. 1996, 271, 17296–17303. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; White, J.A.; Wilson, J.E. Characterization of the rat Type III hexokinase gene promoter. A functional octamer 1 motif is critical for basal promoter activity. J. Biol. Chem. 1999, 274, 31700–31706. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Edassery, S.; Wilson, J.E. The human gene for the Type III isozyme of hexokinase. Structure, basal promoter, and evolution. Arch. Biochem. Biophys. 2001, 395, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Pietilä, M.; Halmekytö, M.; Suppola, S.; Pirinen, E.; Deeb, S.S.; Jänne, J.; Laakso, M. Hexokinase II-deficient mice. Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J. Biol. Chem. 1999, 274, 22517–22523. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kanno, H.; Tancabelic, J.; Fujii, H. Gene expression and biological significance of hexokinase in erythroid cells. Acta Haematol. 2002, 108, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, M.N.; Owens, N.D.L.; Hopkinson, J.R.; Johnson, M.B.; Houghton, J.A.L.; Dasta-mani, A.; Flaxman, C.S.; Wyatt, R.C.; Hewat, T.I.; Hopkins, J.J.; et al. Non-coding variants disrupting a tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. Nat. Genet. 2022, 54, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Feriotto, G.; Finotti, A.; Breveglieri, G.; Treves, S.; Zorzato, F.; Gambari, R. Transcriptional activity and Sp 1/3 transcription factor binding to the P1 promoter sequences of the human AbetaH-J-J locus. FEBS J. 2007, 274, 4476–4490. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, X.; Wang, J.; Xu, L.; Zhang, L.; Xu, B.; Su, J.; Bian, X. LINC00365 functions as a tumor suppressor by inhibiting HIF-1α-mediated glucose metabolism reprogramming in breast cancer. Exp. Cell Res. 2023, 425, 113514. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Roe, J.S.; Lee, J.E.; Cho, E.J.; Youn, H.D. p53 regulates glucose metabolism by miR-34a. Biochem. Biophys. Res. Commun. 2013, 437, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.W.; Nemunaitis, J. Modulation of miRNA activity in human cancer: A new paradigm for cancer gene therapy? Cancer Gene Ther. 2008, 15, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a pro-pensity for mitochondrial binding. J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar] [CrossRef]
- Rempel, A.; Mathupala, S.P.; Griffin, C.A.; Hawkins, A.L.; Pedersen, P.L. Glucose catabolism in cancer cells: Amplification of the gene encoding type II hexokinase. Cancer Res. 1996, 56, 2468–2471. [Google Scholar] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Aberrant glycolytic metabolism of cancer cells: A remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J. Bioenerg. Biomembr. 1997, 29, 339–343. [Google Scholar] [CrossRef]
- Mayer, D.; Klimek, F.; Rempel, A.; Bannasch, P. Hexokinase expression in liver preneoplasia and neoplasia. Biochem. Soc. Trans. 1997, 25, 122–127. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Katzen, H.M. The effect of diabetes and insulin in vivo and in vitro on a low Km form of hexokinase from various rat tissues. Biochem. Biophys. Res. Commun. 1966, 24, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Katzen, H.M.; Soderman, D.D.; Wiley, C.E. Multiple forms of hexokinase. Activities associated with subcellular particulate and soluble fractions of normal and streptozotocin diabetic rat tissues. J. Biol. Chem. 1970, 245, 4081–4096. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Printz, R.L.; Kande, J.; Assan, R.; Granner, D.K.; Girard, J. Regulation of glucose transporter and hexokinase II expression in tissues of diabetic rats. Am. J. Physiol. 1993, 265 Pt 1, E392–E401. [Google Scholar] [CrossRef] [PubMed]
- Gurel, E.; Ustunova, S.; Kapucu, A.; Yilmazer, N.; Eerbeek, O.; Nederlof, R.; Hollmann, M.W.; Demirci-Tansel, C.; Zuurbier, C.J. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol. Biol. Rep. 2013, 40, 4153–4160. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Horton, J.D.; Wilson, J.E. Anabolic function of the Type II isozyme of hexokinase in hepatic lipid synthesis. Biochem. Biophys. Res. Commun. 2000, 270, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Kaselonis, G.L.; McCabe, E.R.; Gray, S.M. Expression of hexokinase 1 and hexokinase 2 in mammary tissue of nonlactating and lactating rats: Evaluation by RT-PCR. Mol. Genet. Metab. 1999, 68, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P.B. Molecular physiology of mammalian glucokinase. Cell Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Moates, J.M.; Nanda, S.; Cissell, M.A.; Tsai, M.-J.; Stein, R. BETA2 activates transcription from the upstream glucokinase gene promoter in islet β-cells and gut endocrine cells. Diabetes 2003, 52, 403–408. [Google Scholar] [CrossRef]
- Jetton, T.L.; Liang, Y.; Pettepher, C.C.; Zimmerman, E.C.; Cox, F.G.; Horvath, K.; Matschinsky, F.M.; Magnuson, M.A. Analysis of up-stream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J. Biol. Chem. 1994, 269, 3641–3654. [Google Scholar] [CrossRef]
- Moates, J.M.; Magnuson, M.A. The Pal elements in the upstream glucokinase promoter exhibit dyad symmetry and display cell-specific enhancer activity when multimerized. Diabetologia 2004, 47, 1632–1640. [Google Scholar] [CrossRef]
- Sternisha, S.M.; Miller, B.G. Molecular and Cellular Regulation of Human Glucokinase. Arch. Biochem. Biophys. [CrossRef]
- Peter, A.; Stefan, N.; Cegan, A.; Walenta, M.; Wagner, S.; Königsrainer, A.; Königsrain-er, A.; Machicao, F.; Schick, F.; Häring, H.-U.; et al. Hepatic Glucokinase Expression Is Associated with Lipogenesis and Fatty Liver in Humans. J. Clin. Endocrinol. Metab. 2011, 96, E1126–E1130. [Google Scholar] [CrossRef]
- Iynedjian, P.B.; Gjinovci, A.; Renold, A.E. Stimulation by insulin of glucokinase gene transcription in liver of diabetic rats. J. Biol. Chem. 1988, 263, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Okar, D.A.; Stoeckman, A.K.; Peng, L.J.; Herrera, A.H.; Herrera, J.E.; Towle, H.C.; Lange, A.J. A potential role for fructose-2,6-bisphosphate in the stimulation of hepatic glucokinase gene expression. Endocrinology 2004, 145, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Roth, U.; Curth, K.; Unterman, T.G.; Kietzmann, T. The transcription factors HIF-1 and HNF-4 and the coactivator p300 are involved in insulin-regulated glucokinase gene expression via the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2004, 279, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P.B.; Marie, S.; Gjinovci, A.; Genin, B.; Deng, S.P.; Buhler, L.; Morel, P.; Mentha, G. Glucokinase and cytosolic phosphoenolpyruvate carboxykinase (GTP) in the human liver. Regulation of gene expression in cultured hepatocytes. J. Clin. Investig. 1995, 95, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Agius, L. Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu. Rev. Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.; Aiston, S.; Agius, L. Subcellular localization, mobility, and kinetic activity of glucokinase in glucose-responsive insulin-secreting cells. Diabetes 2000, 49, 2048–2055. [Google Scholar] [CrossRef]
- Preller A andWilson, J.E. Localization of the type III isozyme of hexokinase at the nuclear periphery. Arch. Biochem. Biophys. 1992, 294, 482–492. [Google Scholar] [CrossRef]
- Guillaume, C.; Bernard, R.; Scott, J.; Paavo, K.; Peipei, P.; James, N.W. Hexokinases and cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 107–115. [Google Scholar]
- Nishizawa, T.; Kanter, J.E.; Kramer, F.; Barnhart, S.; Shen, X.; Vivekanandan-Giri, A.; Wall, V.Z.; Kowitz, J.; Devaraj, S.; O’Brien, K.D.; et al. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. 2014, 7, 356–365. [Google Scholar] [CrossRef]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi AMK. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.; Keyhani-Nejad, F.; Pusec, C.M.; Goodman, L.; Geier, J.A.; Stoolman, J.S.; Stanczyk, P.J.; Nguyen, T.; Xu, K.; Suresh, K.V.; et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell. 2022, 82, 1261–1277.e6. [Google Scholar] [CrossRef] [PubMed]
- van der Kooij, M.A.; Rojas-Charry, L.; Givehchi, M.; Wolf, C.; Bueno, D.; Arndt, S.; Ten-zer, S.; Mattioni, L.; Treccani, G.; Hasch, A.; et al. Chronic social stress disrupts the intracellular redistribution of brain hexokinase 3 induced by shifts in peripheral glucose levels. J. Mol. Med. 2022, 100, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.B.; Kindzelskii, A.L.; Petty, H.R. Hexokinase translocation during neutrophil activation, chemotaxis, and phagocytosis: Disruption by cytochalasin, D.; dexamethasone, and indomethacin. Cell. Immunol. 2002, 218, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Brown, L.G.; Coleman, I.M.; Nguyen, H.M.; Lin, D.W.; Corey, E.; Nelson, P.S.; Morrissey, C. Cabozantinib can block growth of neuroendocrine prostate cancer patient-derived xenografts by disrupting tumor vasculature. PLoS ONE 2021, 16, e0245602. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Kim, H.; Son, T.; Jeong, Y.; Kim, S.U.; Dong, S.M.; Park, Y.N.; Lee, J.D.; Lee, J.M.; Park, J.H. Regulation of HK2 expression through alterations in CpG methylation of the HK2 promoter during progression of hepatocellular carcinoma. Oncotarget 2016, 7, 41798–41810. [Google Scholar] [CrossRef]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and, II. Mol. Cell Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E. Regulation of mammalian hexokinase activity. In Regulation of Carbohydrate Metabolism; Beitner, R., Ed.; CRC: Boca Raton, FL, USA, 1985; pp. 45–85. [Google Scholar]
- Li, Y.; Tian, H.; Luo, H.; Fu, J.; Jiao, Y.; Li, Y. Prognostic significance and related mechanisms of hexokinase 1 in ovarian cancer. Onco Targets Ther. 2020, 13, 11583–11594. [Google Scholar] [CrossRef]
- Xu, S.; Catapang, A.; Doh, H.M.; Bayley, N.A.; Lee, J.T.; Braas, D.; Graeber, T.G.; Herschman, H.R. Hexokinase 2 is targetable for HK1 negative, HK2 positive tumors from a wide variety of tissues of origin. J. Nucl. Med. 2019, 60, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Catapang, A.; Braas, D.; Stiles, L.; Doh, H.M.; Lee, J.T.; Graeber, T.G.; Damoiseaux, R.; Shirihai, O.; Herschman, H.R. A precision therapeutic strategy for hexokinase 1-null, hexokinase 2-positive cancers. Cancer Metab. 2018, 6, 7. [Google Scholar] [CrossRef]
- Šimčíková, D.; Gardáš, D.; Hložková, K.; Hruda, M.; Žáček, P.; Rob, L.; Heneberg, P. Loss of hexokinase 1 sensitizes ovarian cancer to high-dose metformin. Cancer Metab. 2021, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Amendola, C.R.; Mahaffey, J.P.; Parker, S.J.; Ahearn, I.M.; Chen, W.C.; Zhou, M.; Court, H.; Shi, J.; Mendoza, S.L.; Morten, M.J.; et al. KRAS4 directly regulates HK1. Nature 2019, 576, 482–486. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour Biol. 2015, 36, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, M.; Sun, H.; Wang, F.; Xie, X.; Chen, X.; Su, J.; He, Y.; Dai, Y.; Wu, H.; et al. HK2 is a radiation resistant and independent negative prognostic factor for patients with locally advanced cervical squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 4054–4063. [Google Scholar]
- Iwamoto, M.; Kawada, K.; Nakamoto, Y.; Itatani, Y.; Inamoto, S.; Toda, K.; Kimura, H.; Sasazuki, T.; Shirasawa, S.; Okuyama, H.; et al. Regulation of ^18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J. Nucl. Med. 2014, 55, 2038–2044. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Marybeth, A.; Raoud, M.; Richard, M.; Jen, J.Y. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2016, 8, 56081–56094. [Google Scholar]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef]
- Neary, C.L.; Pastorino, J.G. Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J. Cell Physiol. 2013, 228, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Li, M.; Liu, W.B.; Zhou, Z.S.; Zhang, R.; Li, J.L.; Zhou, K.C. Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2-mediated glycolysis. Oncol. Rep. 2015, 33, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Liu, Y.; Liu, H.; Chen, X.; Liu, M.; Che, H.; Guo, F.; Wang, C.; Zhang, D.; Wu, J.; et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci. Rep. 2015, 5, 9065. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Enokida, H.; Itesako, T.; Kojima, S.; Kinoshita, T.; Tatarano, S.; Chiyomaru, T.; Nakagawa, M.; Seki, N. Tumor-suppressive microRNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer Sci. 2013, 104, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Qiu, Z.; Wang, Z.; Wang, Q.; Tan, N.; Chen, T.; Chen, Z.; Huang, S.; Gu, J.; Li, J.; et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology 2015, 62, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Cheng, C.; Lu, H.; Wang, Y. miR-4458 suppresses glycolysis and lactate production by directly targeting hexokinase2 in colon cancer cells. Biochem. Biophys. Res. Commun. 2016, 469, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 232. [Google Scholar] [CrossRef]
- Dai, W.; Wang, F.; Lu, J.; Xia, Y.; He, L.; Chen, K.; Li, J.; Li, S.; Liu, T.; Zheng, Y.; et al. By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget 2015, 6, 13703–13717. [Google Scholar] [CrossRef]
- Federzoni, E.A.; Valk, P.J.; Torbett, B.E.; Haferlach, T.; Löwenberg, B.; Fey, M.F.; Tschan, M.P. PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 2012, 119, 4963–4970. [Google Scholar] [CrossRef] [PubMed]
- Hai-Yan, G.; Xin-Guo, L.; Xi, C.; Jing-Hua, W. Identification of key genes affecting disease free survival time of pediatric acute lymphoblastic leukemia based on bioinformatic analysis. Blood Cells Mol. Dis. 2015, 54, 38–43. [Google Scholar]
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Seiler, K.; Humbert, M.; Minder, P.; Mashimo, I.; Schläfli, A.M.; Krauer, D.; Federzoni, E.A.; Vu, B.; Moresco, J.J.; Yates, J.R., 3rd; et al. Hexokinase 3 enhances myeloid cell survival via non-glycolytic functions. Cell Death Dis. 2022, 13, 448. [Google Scholar] [CrossRef]
- Xu, W.; Liu, W.R.; Xu, Y.; Tian, X.; Anwaier, A.; Su, J.Q.; Zhu, W.K.; Shi, G.H.; Wei, G.M. Hexokinase 3 dysfunction promotes tumorigenesis and immune escape by upregulating monocyte/macrophage infiltration into the clear cell renal cell carcinoma microenvironment. Int. J. Biol. Sci. 2021, 17, 2205–2222. [Google Scholar] [CrossRef] [PubMed]
- Board, M.; Colquhoun, A.; Newsholme, E.A. High Km glucose-phosphorylating (glucokinase) activities in a range of tumor cell lines and inhibition of rates of tumor growth by the specific enzyme inhibitor mannoheptulose. Cancer Res. 1995, 55, 3278–3285. [Google Scholar] [PubMed]
- Bui, N.L.C.; Pandey, V.; Zhu, T.; Ma, L.; Basappa, L.P.E. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018, 415, 17–26. [Google Scholar] [CrossRef]
- Danial, N.N. BAD: Undertaker by night, candyman by day. Oncogene 2008, 27 (Suppl. 1), S53–S70. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Heese, K.; Wu, M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol. Cell Biol. 2006, 26, 9071–9082. [Google Scholar] [CrossRef]
- Matschinsky, F.M.; Magnuson, M.A.; Zelent, D.; Jetton, T.L.; Doliba, N.; Han, Y.; Taub, R.; Grimsby, J. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 2006, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.; Ramakanth, S.; Wendy, L.C.; Nancy-Ellen, H.; Fred, T.B.; John, W.C.; Kevin, R.G.; Darryl, H.; Robert, K. Allosteric Activators of Glucokinase: Potential Role in Diabetes Therapy. Science 2003, 301, 370–373. [Google Scholar]
- Matschinsky, F.M. Assessing the potential of glucokinase activators in diabetes therapy. Nat. Rev. Drug Discov. 2009, 8, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Akinobu N and Yasuo, T. Present status of clinical deployment of glucokinase activators. J. Diabetes Investig. 2015, 6, 124–132. [Google Scholar]
- Yoon, S.O.; Youn-Jung, L.; Kaapjoo, P.; Hyun Ho, C.; Sangjong, Y.; Hee-Sook, J. Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells. Eur. J. Pharm. Sci. 2014, 51, 137–145. [Google Scholar]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Kassem, S.; Bhandari, S.; Rodríguez-Bada, P.; Motaghedi, R.; Heyman, M.; García-Gimeno, M.A.; Cobo-Vuilleumier, N.; Sanz, P.; Maclaren, N.K.; Rahier, J.; et al. Large islets, beta-cell proliferation, and a glucokinase mutation. N. Engl. J. Med. 2010, 362, 1348–1350. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Cheng, F.; Song, H.; Lu, W.; Zhao, J.; An, X.; Liu, M.; Chen, G.; Zhao, Z.; Zhang, J. Proteome-Scale Investigation of Protein Allosteric Regulation Perturbed by Somatic Mutations in 7,000 Cancer Genomes. Am. J. Hum. Genet. 2017, 100, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Těšínský, M.; Šimčíková, D.; Heneberg, P. First evidence of changes in enzyme kinetics and stability of glucokinase affected by somatic cancer-associated variations. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 213–218. [Google Scholar] [CrossRef]
- Orci, L.A.; Sanduzzi-Zamparelli, M.; Caballol, B.; Sapena, V.; Colucci, N.; Torres, F.; Bruix, J.; Reig, M.; Toso, C. Incidence of hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Clin. Gastroenterol. Hepatol. 2021, 20, 283–292.e10. [Google Scholar] [CrossRef]
- Nagaoki, Y.; Hyogo, H.; Ando, Y.; Kosaka, Y.; Uchikawa, S.; Nishida, Y.; Teraoka, Y.; Morio, K.; Fujino, H. Increasing incidence of non-HBV- and non-HCV-related hepatocellular carcinoma: Single-institution 20-year study. BMC Gastroenterol. 2021, 21, 306. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, Y.; Yang, L.; Chen, S. A prognostic 4-gene expression signature for squamous cell lung carcinoma. J. Cell. Physiol. 2017, 232, 3702–3713. [Google Scholar] [CrossRef]
- Yixiang, Z.; Puyuan, X.; Junling, L. Treatment of advanced squamous cell lung cancer. Chin. J. Lung Cancer 2016, 19, 687–691. [Google Scholar]
- Jia, H.; Wang, A.; Lian, H.; Shen, Y.; Wang, Q.; Zhou, Z.; Zhang, R.; Li, K.; Liu, C. Identification of novel alternative splicing isoform biomarkers and their association with overall survival in colorectal cancer. BMC Gastroenterol. 2020, 20, 1–12. [Google Scholar]
- Majem, M.; Juan, O.; Insa, A.; Reguart, N.; Trigo, J.M.; Carcereny, E.; García-Campelo, R.; García, Y.; Guirado, M.; Provencio, M. SEOM clinical guidelines for the treatment of non-small cell lung cancer (2018). Clin Transl Oncol. 2019, 21, 3–17. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Fuhr, L.; El-Athman, R.; Scrima, R.; Cela, O.; Carbone, A.; Knoop, H.; Li, Y.; Hoffmann, K.; Laukkanen, M.O.; Corcione, F.; et al. The Circadian Clock Regulates Metabolic Phenotype Rewiring Via HKDC1 and Modulates Tumor Progression and Drug Response in Colorectal Cancer. EBioMedicine 2018, 33, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Huang, C.; Li, J.; Gao, T.; Lin, Z.; Yao, T. Long non-coding RNA urothelial cancer associated 1 regulates radioresistance via the hexokinase 2/glycolytic pathway in cervical cancer. Int. J. Mol. Med. 2018, 42, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hu, M.; Zhou, L.; Ling, S.; Li, Y.; Kong, B.; Huang, P. Long non-coding RNA HOTAIR promotes cancer cell energy metabolism in pancreatic adenocarcinoma by upregulating hexokinase-2. Oncol. Lett. 2019, 18, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Nabi, K.; Le, A. The Intratumoral Heterogeneity of Cancer Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 149–160. [Google Scholar] [CrossRef]
- Antonio, M.J.; Zhang, C.; Le, A. Different Tumor Microenvironments Lead to Different Metabolic Phenotypes. Adv. Exp. Med. Biol. 2021, 1311, 137–147. [Google Scholar] [CrossRef]
- Evstafieva, A.G.; Kovaleva, I.E.; Shoshinova, M.S.; Budanov, A.V.; Chumakov, P.M. Implication of KRT16, FAM129A and HKDC1 genes as ATF4 regulated components of the integrated stress response. PLoS ONE 2018, 13, e0191107. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.N.; Yu, B.B.; Li, J.L.; Guo, R.; Zhang, L.C.; Sun, L.K.; Liu, Y.N.; Li, Y. Zinc and p53 disrupt mitochondrial binding of HK2 by phosphorylating VDAC1. Exp. Cell Res. 2019, 374, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhuang, Y.; Xu, J.; Tong, Y.; Li, X.; Dong, C. Advances in the Study of Hexokinase 2 (HK2) Inhibitors. Anticancer Agents Med. Chem. 2023, 23, 736–746. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HK1 | HK2 | HK3 | GCK | HKDC1 | |
|---|---|---|---|---|---|
| Gene location (Human) | 10q22 | 2p13 | 5q35.2 | 7p15.1 | 10q22 |
| MW (kDa) | ~100 | ~100 | ~100 | ~50 | ~100 |
| Number of catalytic domains | 1 | 2 | 1 | 1 | 1 |
| Km for glucose (mmol L−1) | 0.03 | 0.3 | 0.003 | 6 | - |
| Km for ATP (mmol L−1) | 0.5 | 0.7 | 1.0 | 0.6 | - |
| G6P inhibition Ki (mmol L−1) | 0.02 | 0.02 | 0.10 | - | - |
| Effect of Pi | Low conc counteracts G6P inhibition, but high conc is inhibitory | inhibitory | Inhibitory | - | - |
| Insulin regulation | - | + | * | + | * |
| Major tissue expression | Brain, Kidney | Muscle, adipose | Lung, spleen | Liver, pancreas | GI, Kidney, and Brain |
| Mitochondrial binding | ✓ | ✓ | ✕ | ✕ | ✓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, Z.; Ismail, H.; Bhat, S.A.; Layden, B.T.; Khan, M.W. Aiding Cancer’s “Sweet Tooth”: Role of Hexokinases in Metabolic Reprogramming. Life 2023, 13, 946. https://doi.org/10.3390/life13040946
Farooq Z, Ismail H, Bhat SA, Layden BT, Khan MW. Aiding Cancer’s “Sweet Tooth”: Role of Hexokinases in Metabolic Reprogramming. Life. 2023; 13(4):946. https://doi.org/10.3390/life13040946
Chicago/Turabian StyleFarooq, Zeenat, Hagar Ismail, Sheraz Ahmad Bhat, Brian T. Layden, and Md. Wasim Khan. 2023. "Aiding Cancer’s “Sweet Tooth”: Role of Hexokinases in Metabolic Reprogramming" Life 13, no. 4: 946. https://doi.org/10.3390/life13040946
APA StyleFarooq, Z., Ismail, H., Bhat, S. A., Layden, B. T., & Khan, M. W. (2023). Aiding Cancer’s “Sweet Tooth”: Role of Hexokinases in Metabolic Reprogramming. Life, 13(4), 946. https://doi.org/10.3390/life13040946